Signalment:
One-month-old
female Taconic line 8440 mouse (
Mus musculus).Four mice were
presented dead. Three were found dead and one was euthanized prior to
submission. According to the history, they were treated with azoxymethane.
Gross Description:
All
mice have congested livers with light brown apical margins. Heart and
kidneys are pale. The mice that were euthanized had blood tinged intestinal
contents.
Histopathologic Description:
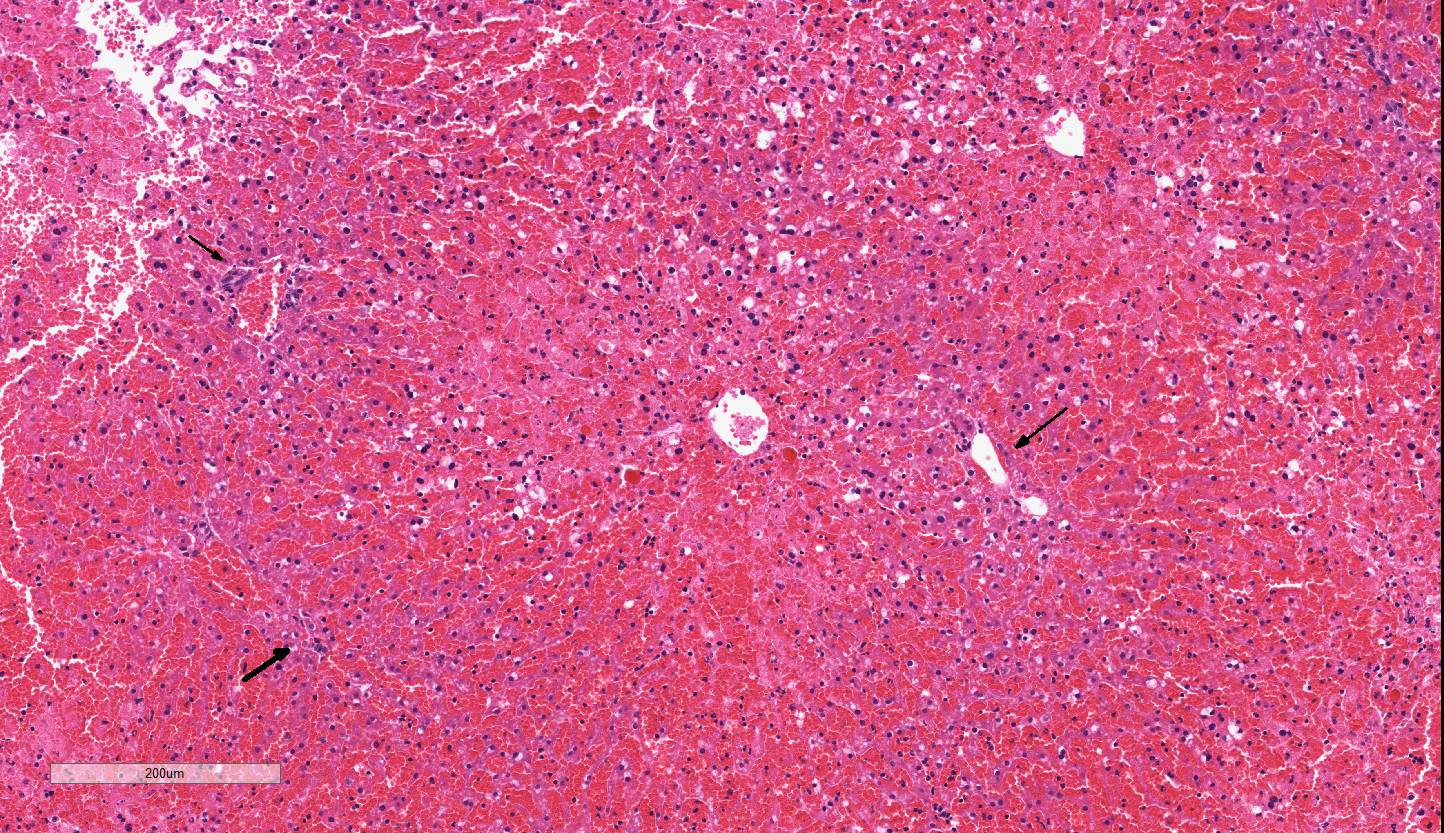
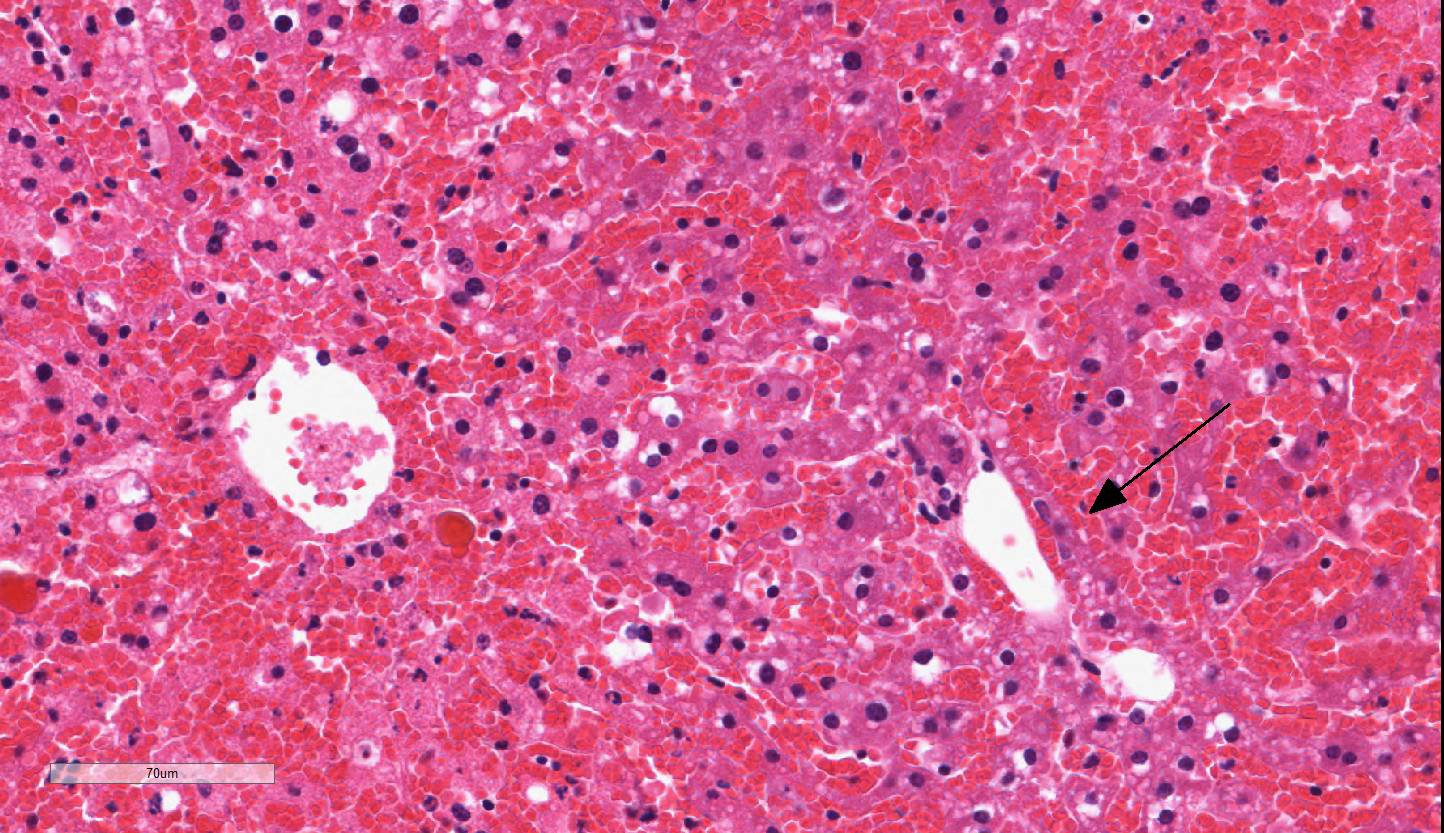
The liver has severe congestion of hepatic lobules with attenuation and loss of
centrilobular hepatocytes. Portal zone hepatocytes are spared but have variably
vacuolated cytoplasm. Other histology
findings include lymphocytolysis of the thymus. The mouse with intestinal
hemorrhage has gastric ulcers. One mouse has necrosis of the adrenal cortex.
Morphologic Diagnosis:
Liver:
Centrilobular to mid-zonal necrosis,
and hemorrhage, severe, acute; micro-vesicular lipidosis mild to moderate.
Lab Results:
Bacteriology
:
Small Intestine:
Enterococcus faecalis,
Staphylococcus sciuri,
and
E. coli; Negative for strict anaerobes.
Condition:
Azoxymethane toxicity
Contributor Comment:
Azoxymethane
(AOM) is derived from the cycad palm nut of Guam. Ingestion has been
associated with acute liver failure in cattle
.1
In
mice and rats, it is used as a mutagen in the study of colon carcinogenesis and
liver failure. It is an alkylating agent that forms 06-methylguanine (0
6meG)
adducts in DNA. 0
6meG formation has been found in human colon
cancers. In the liver, AOM is oxidized to methylazoxymethanol by cytochrome
P450 2E1 and transported to the colon where it methylates DNA which can cause
G:C to A:T transition mutations.
2,3 Methylazoxymethanol is also
associated with acute and chronic liver toxicity.
4
At reported
doses of 100 ug/g in mice AOM causes acute liver injury. It is characterized
by hepatic necrosis and microvesicular lipidosis. The mechanism of action may
be interference with beta-oxidation of fatty acids in the mitochondria
1 Hemorrhage
is an indication of damage to sinusoidal endothelial cells.
4 Hepatic
necrosis then progresses to liver failure and hepatic encephalopathy.
Acute liver
failure is thought to lead to death via neurologic effects and hypotensive
shock with multi-organ failure. Neurologic effects were thought to be a result
of ammonia and other metabolites directly causing hepatic encephalopathy. More
recent work has shown that inflammation-associated cytokine release contributes
to brain edema and other clinical signs. Acute liver failure can result in
cytokine storms with increased TNF-α, IL-1b, Il-6, and Il-12, There is
also evidence of impaired neutrophil phagocytic activity and increased the risk
of sepsis. Portal hypertension may cause increased bacterial translocation
5
cross the gut.
5, 6, 7
JPC Diagnosis:
Liver: Necrosis
and hemorrhage, centrilobular to midzonal, acute, diffuse, severe with
microvesicular lipidosis, Taconic line 8440 mouse,
Mus musculus
Conference Comment:
Despite some minor slide variability, the contributor
provides a good example of the relatively stereotypical histologic changes
associated with acute toxic hepatic injury. The liver is particularly
susceptible to toxic injury due to constant exposure to ingested chemicals
through the portal blood.
4 Additionally, hepatocytes are responsible
for the metabolism of most endogenous and exogenous substances, by a process
called biotransformation. This process is broken down into three phases based
on the hepatic enzymes involved. Phase I reactions involve oxidation,
reduction, hydrolysis, cyclization, and decyclization of the compound via
cytochrome P450 monooxygenases (CYP) utilizing NADPH and oxygen in the smooth
endoplasmic reticulum of hepatocytes. Phase II involves conjugation of the
metabolite produced in phase I via glucuronidation, sulphation, acetylation, or
methylation, ultimately resulting in a water soluble metabolite that is then
excreted through urine or bile. Phase III reactions involve transporting the
conjugated substances through the hepatocyte and into the bile canaliculus.
4,5,7
Conference participants discussed the various mechanisms
of hepatotoxic liver injury, which are divided into six categories based on the
mechanism of action and cellular targets of the toxin.
4 The
most common mechanism involves biotransformation of indirect-acting toxins by the
CYP system, whichresults in bioactivated toxic metabolites that disrupt
intracellular enzymatic pathways. CYP is abundant in the microsomes of the
smooth endoplasmic reticulum in centrilobular zones which explains the
prevalence of centrilobular to midzonal necrosis in some toxic hepatopathies, including
this case of azoxymethane toxicosis.
1,4,6 There is also inhibition
of hepatic mitochondrial function, thus limiting beta-oxidation of fat and ATP
generation via oxidative phosphorylation. This inhibition eventually leads to
necrosis (due to production of damaging reactive oxygen species and lactic
acid), as well as hepatocellular microvesicular lipid accumulation, as seen
adjacent to areas of necrosis in this case.
4 Readers are encouraged
to review 2012
WSC Conference 18 Case 2 for further discussion
of other mechanisms of hepatotoxic liver injury.
In addition to hepatic necrosis, acute and fatal
hepatotoxicity causes destruction of the endothelium of the sinusoidal lining
cells, resulting in zonal areas of hemorrhage. Widespread hemorrhage is often
associated with acute hepatocellular toxicity due to consumption of platelets
and decreased production of clotting factors by the liver.
4
Centrilobular necrosis is the most common form of zonal
hepatocellular necrosis observed in animals and may be caused by a variety of
infectious, inflammatory, metabolic, and toxic insults. Although conference participants could not
identify azoxymethane as the cause of the lesions in this mouse, most suspected
a toxic etiology based upon the presence of centrilobular necrosis and
hemorrhage combined with the lack of evidence of an infectious etiology.
References:
1. Belanger M, et
al. Neurobiological characterization of an azoxymethane mouse model of acute
liver failure.
Neurochem Int. 2006; (48):434-440.
2. Nyskolus LS, et
al. Repair and removal of azoxymethane-induced 0
6-methyguanine in
rat colon by0
6-methyguanine DNA methyltransferase and apoptosis.
Mutat
Res. 2013; 758: 80-86.
3. Sohn OS, et al.
Metabolism of azoxymethane, methylazoxymethanol and N-nitrosodimethylamine by
cytochrome p450IIE1.
Carcinogenesis. 1991; 12 (1):127-131.
4. Stalker MJ,
Cullen JM. Liver and biliary system, In: Maxie MG, ed.
Jubb, Kennedy and
Palmer´s. Pathology of Domestic Animals. Vol 2. 6th ed. St Louis, MO: Elsevier
Saunders; 2016:325-330.
5. Tranah TH, et
al. Systemic inflammation and ammonia in hepatic encephalopathy.
Metab
Brain Dis. 2013; 28:1-5.
6. Bemeur C, Desjardins
P, Butterworth RF. Antioxidant and anti-inflammatory effects of mild
hypothermia in the attenuation of liver injury due to azoxymethane toxicity in
the mouse.
Metab Brain Dis. 2010; 25:23-29.
7. Chastre A, et
al. Inflammatory cascade driven by tumor necrosis factor-alpha play a major
role in the progression of acute liver failure and its neurologic
complications.
PLoS One. 2012; 7(11):e49670.