Signalment:
Eight-year-old,
female, Labrador retriever, (
Canis familiaris).The dog had a
history of polyuria and polydipsia with several urinary accidents. Otherwise,
she was clinically healthy. Initial blood work showed azotemia (BUN: 26 mg/dL
[reference range: 5-20]; serum creatinine: 2.0 mg/dL [reference range:
0.6-1.6]) and glucosuria (4+) with normal blood glucose. There was also
increased ALT (458 IU/L [reference range: 10-55]). Her urine was isosthenuric
and contained fine granular casts. Her blood pressure was normal (systemic
blood pressure: 140 mmHg). There was no history of exposure to toxins.
Subsequent blood work one month later showed increased azotemia (BUN: 23 mg/dL;
serum creatinine: 3.0 mg/dL), ALT (707 IU/L), and mild proteinuria (UPC: 1.3
[normal <0.5]).
Gross Description:
A wedge
biopsy of the renal cortex was submitted for evaluation. Wedge biopsies of
liver were also harvested and submitted to a different diagnostic laboratory.
No gross abnormalities were observed at surgery.
Histopathologic Description:
(H&E):
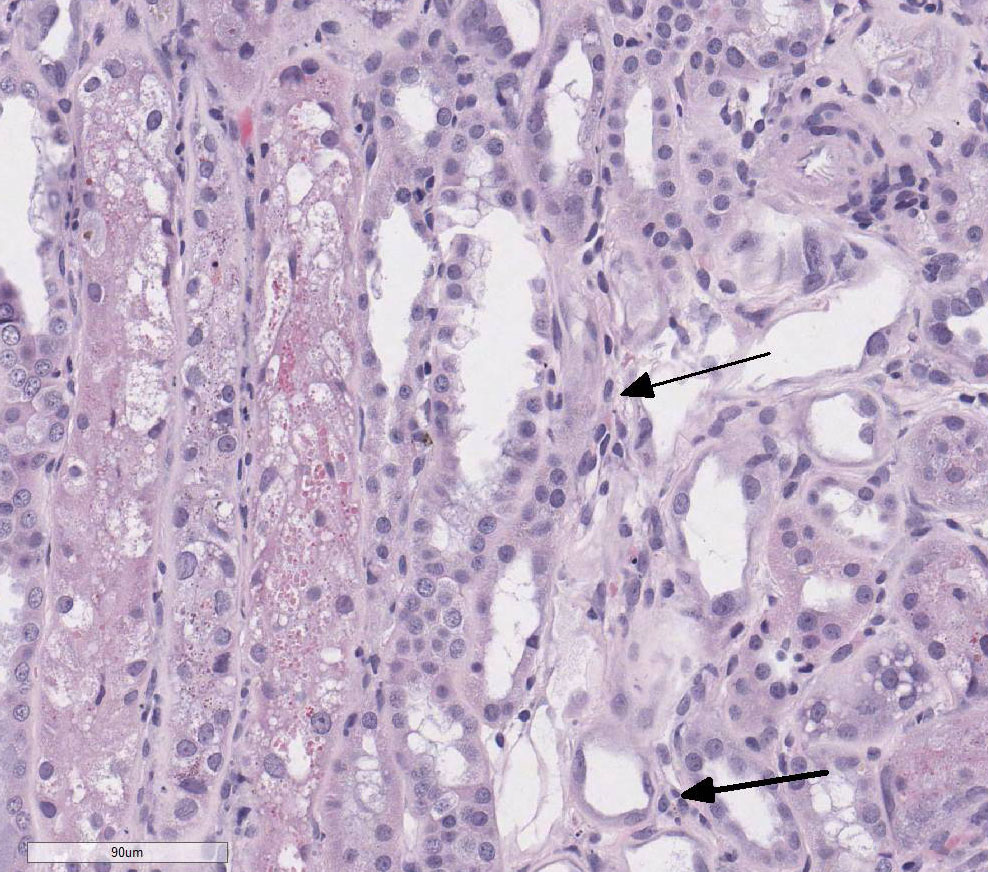
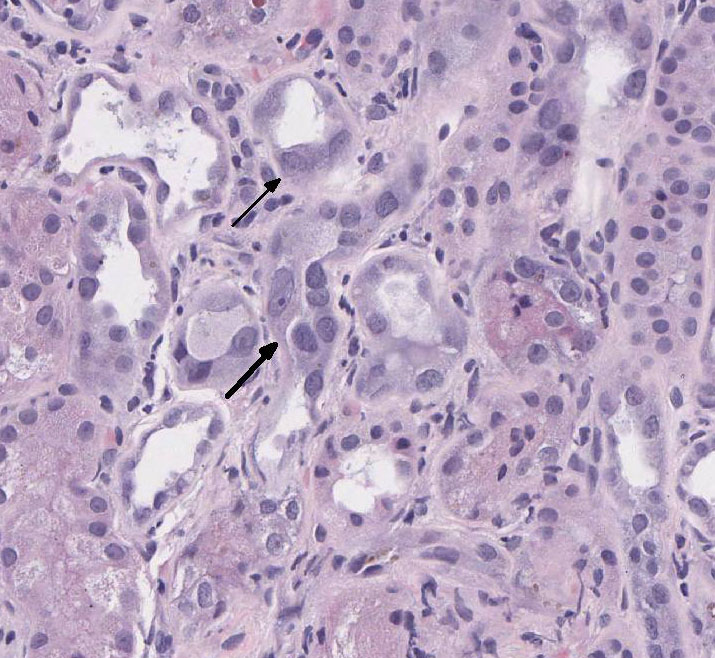
There is loss of the apical brush border of the proximal tubules. Scattered
tubules are necrotic with cellular casts within lumens and attenuated or absent
epithelial lining. Tubules are dilated and undergoing degeneration with single
cell necrosis and apoptosis. The tubular epithelial cells contain variably
sized cytoplasmic vacuoles with abundant granular pigment. There is marked
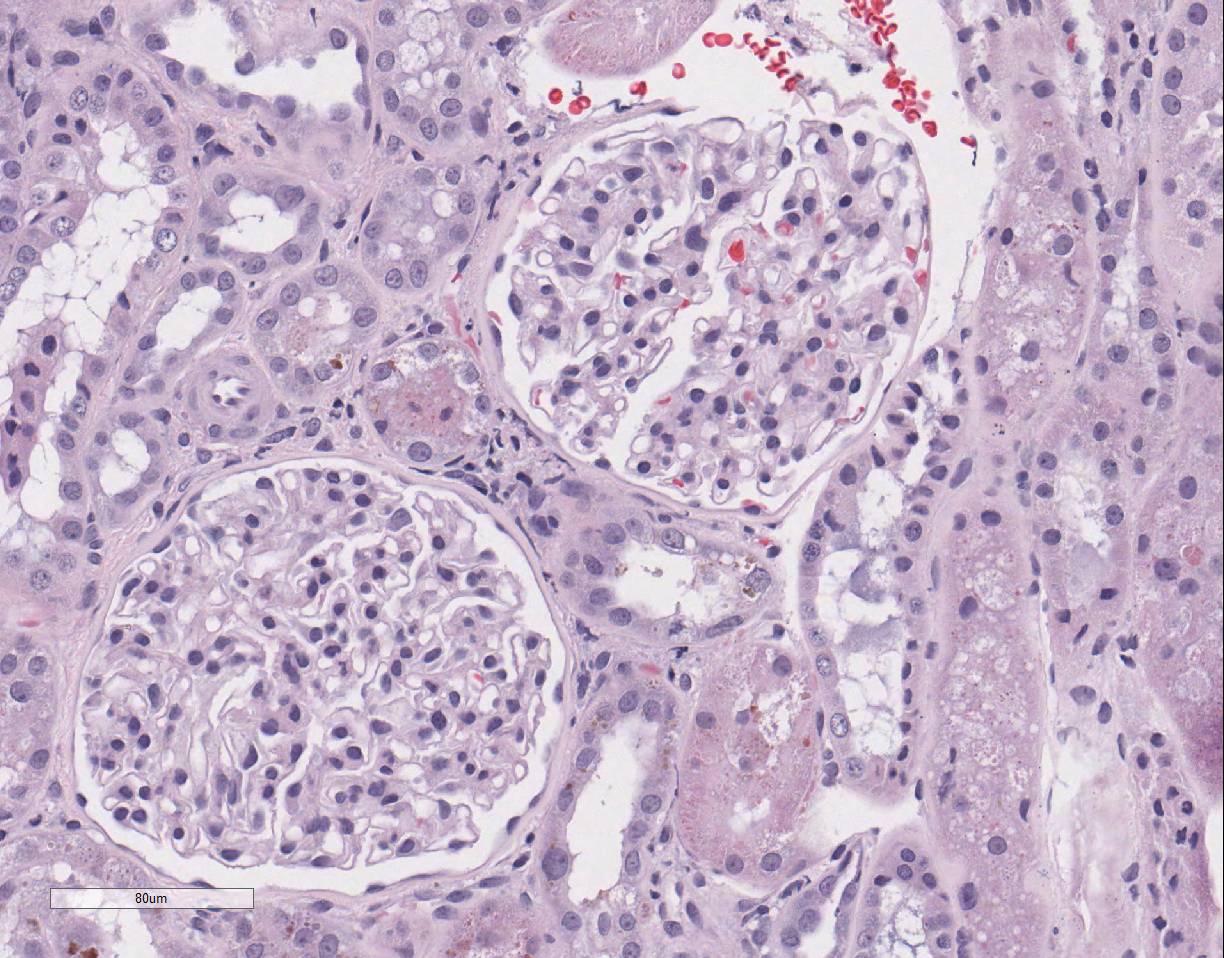
epithelial cell karyomegaly. A few glomeruli
have minimal to mild mesangial expansion and the interstitium is mildly
expanded by fibroplasia with scattered aggregates of lymphocytes and
macrophages.
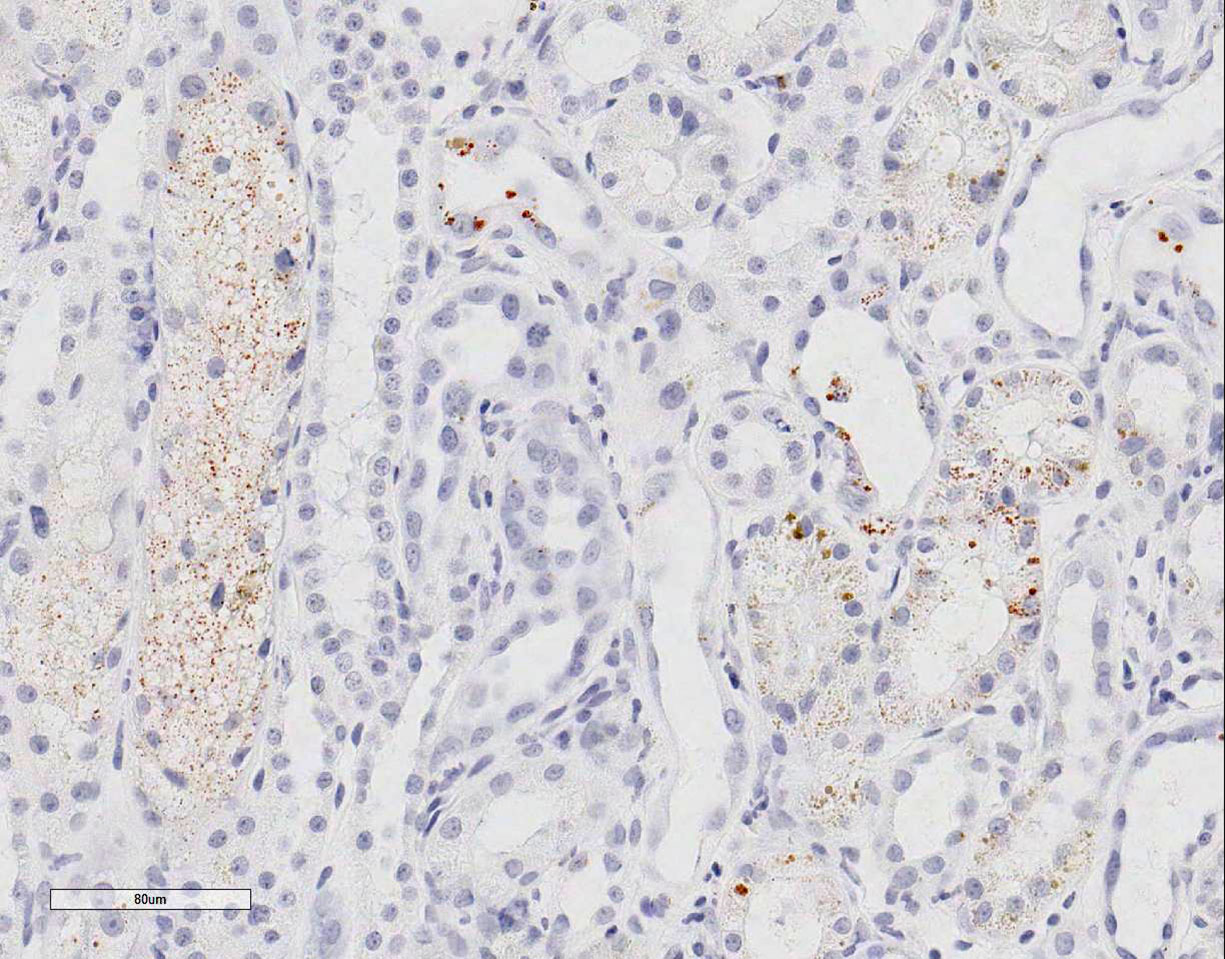
Special stains: Rhodanine stain:
Scattered tubular epithelial cells contain small and large red-brown granules,
consistent with copper. PAS demonstrates multifocal marked loss of the apical
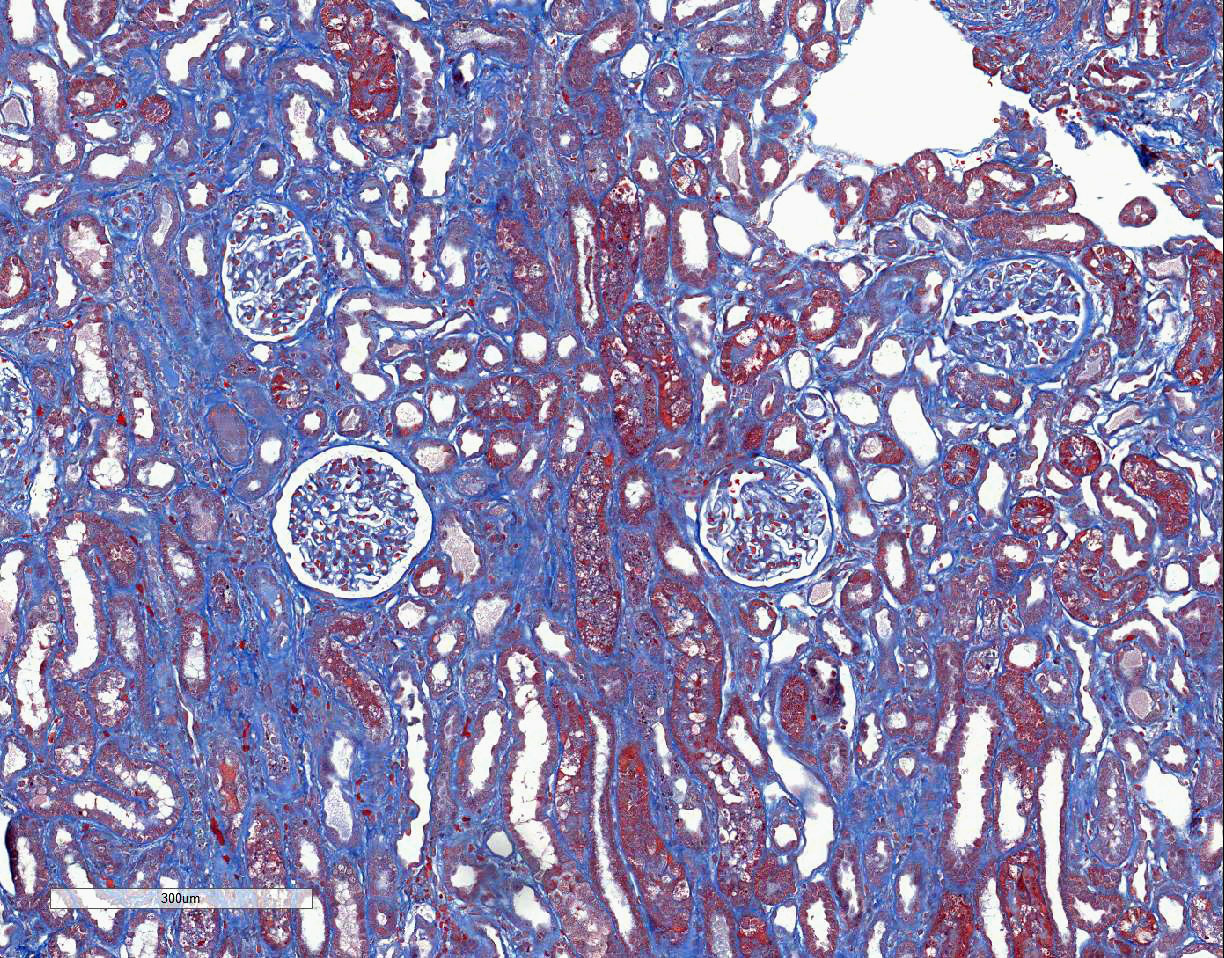
brush borders. The trichrome stain demonstrates a few large regions of mild to
moderate interstitial fibrosis.
Morphologic Diagnosis:
Moderate to severe tubular degeneration with necrosis, regeneration, atrophy,
epithelial cell karyomegaly and scattered intracytoplasmic copper within
tubular epithelial cells. Mild multifocal interstitial fibrosis.
Lab Results:
The unfixed
renal tissue that had been submitted for IF evaluation, and a portion of the
liver sample were submitted to Colorado State Diagnostic Laboratory to measure
copper. There was evidence of copper hepatopathy (copper 2,690 ppm [>1,500
ppm in the liver is considered toxic]) and copper in the kidney 243.00 ppm
(relevant reference range >100 ppm in non-hepatic tissue being indicative of
toxicity). Urine was also submitted approximately 10 days after the biopsy.
SDS-PAGE analysis of urine demonstrated proteinuria due to the presence of low
molecular weight proteins, consistent with tubular damage and absence of
glomerular injury. An aliquot of this urine sample was submitted to PennGen
which documented generalized amino-aciduria and glucosuria without ketonuria or
cystinuria warranting a diagnosis of acquired Fanconi syndrome.
Condition:
Fanconi-like syndrome
Contributor Comment:
The lesions presented in this case are indicative of renal proximal tubular
injury, which was clinically supported by the urinalysis results. These
acquired lesions have been associated with copper storage hepatopathy as a part
of Wilsons disease in humans and dogs. Acquired Fanconi syndrome is
characterized by impaired reabsorptive function of the proximal renal tubules.
Clinical features include excessive loss of water, glucose, amino acids, uric
acid in the urine, and electrolyte abnormalities. The inherited form of
Fanconi-like syndrome is well described in Basenji dog and is thought to be due
to increased amounts of cholesterol in the tubular epithelial brush border
compared to normal dogs. In contrast, acquired proximal renal tubulopathies
have been loosely characterized in the literature and is attributed to many
causes including copper hepatopathy, leptospirosis, hypo-parathyroidism,
ethylene glycol toxicity, antibiotic, and chicken jerky treats.
In
humans, Wilsons disease is an autosomal recessive disorder of copper
metabolism caused by mutations in the
ATP7B gene. Decreased expression
of this gene leads to decreased biliary excretion of copper resulting in
hepatic copper accumulation. Patients with Wilsons disease also accumulate
copper in various tissues including the brain, eye, and kidney. Copper storage
diseases have been reported in several canine breeds including Bedlington
terrier, Labrador retriever, Doberman pinscher, Dalmatian, Skye terrier and
West Highland white terrier. The inherited form of copper storage disease has
been well documented in Bedlington terrier in which the copper metabolism
domain containing 1 (
COMMD1) gene is affected. The COMMD1 protein is
important for copper excretion into bile during states of elevated
intracellular copper. Hepatic histopathology of copper associated hepatopathies
generally present with mixed inflammation (neutro-philic, lymphoplasmacytic,
histiocytic) and is usually localized to the centrilobular regions.
Centrilobular necrosis, bridging fibrosis, and cirrhosis have also been
described in copper-associated hepatitis. Of note, chronic liver injury will
lead to increased amounts of copper in the hepatocytes, sometimes making it
difficult to discern whether intracellular copper is the cause or the effect of
the liver injury.
A few case
series/reports have documented acquired proximal renal tubulopathies associated
with copper storage hepatopathy in dogs. Breeds included are: Clumber spaniel,
West Highland white terrier, Cardigan Welsh corgi, and Labrador retriever. The
renal histologic findings in previous reported cases were consistent with the
case presented here. Histologic evaluation of the renal tissues showed proximal
tubular epithelial degeneration, necrosis, and regeneration. The tubular
epithelial cells were plump with variably sized vacuoles. Using rhodanine
stain, one case reported copper deposition was mainly localized to the
corticomedullary junction and medullary areas; however copper staining can be
variable. Therefore, mild tubular epithelial cell degeneration and loss of the
apical brush border in the setting of clinical symptoms of Fanconi syndrome
should alert the pathologist to possible copper-mediated damage to the renal
proximal tubules. Assay of copper levels in the liver or kidney samples
supports this pathogenesis.
In previous case
reports of copper hepatopathy induced proximal renal tubular disease, there was
improvement and resolution of clinical signs with copper chelation therapy,
specifically using d-penicillamine. Additional therapies including supportive
care, antioxidants, and low copper diet can also contribute to improvement of
clinical signs.
JPC Diagnosis:
Kidney,
tubules: Epithelial degeneration, re-generation, and necrosis, diffuse, marked,
with karyomegaly, few tubular casts, intracytoplasmic pigment and interstitial
fibrosis, Labrador retriever,
Canis familiaris.
Conference Comment:
The contributor provides an
excellent example and thorough review of copper-associated acquired
Fanconi-like syndrome. This syndrome is characterized by polyuria, polydipsia,
hyposthenuria, glucosuria with normo-glycemia, hyperphosphaturia, proteinuria,
and amino aciduria due to impaired renal tubular absorption of glucose,
phosphates, sodium, potassium, uric acid, and amino acids.
1,2,4,7 In
this case, this animal had glucosuria with normoglycemia and amino-aciduria
indicating poor proximal convoluted tubular functioning. Glucose is normally
resorbed in the renal proximal tubules via the sodium-glucose co-transport
system.
1,7,9 The concentration gradient established by this system
also promotes sodium resorption from the tubular fluid. In congenital or
acquired tubular defects of Fanconi-like syndrome, glucose is not resorbed and
will cause an osmotic diuresis. This diuresis causes a marked decrease in
kidneys ability to concentrate urine and will increase urine volume; indicated
by polyuria and isosthenuria reported in this case.
1,7,9
The polydipsia is likely secondary to compensation from increased fluid loss in
the urine.
Conference participants discussed the significance of the
reported presence of low molecular weight proteins in the urine using sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). In general,
there are four main types of proteinuria: pre-renal, glomerular, tubular, and
hemorrhagic/inflammatory. In pre-renal proteinuria, small proteins such as
hemoglobin dimers, myoglobin, and light chains are present in the plasma at
increased concentrations and pass through the glomerulus and are incompletely
resorbed by fully functioning tubules.
8 Glomerular proteinuria is
characterized by damage to the glomerulus, thus enabling high molecular weight
and negatively charged proteins to leak into the filtrate and pass into the urine
due to loss of selective permeability.
8 In tubular proteinuria,
proximal renal tubules are damaged or defective so low molecular weight
proteins, like smaller globulins and some albumen, do not get resorbed from the
ultra-filtrate and are excreted in the urine. Hemorrhagic/inflammatory
proteinuria occurs due to hemorrhage or inflammation within the renal tubules,
renal pelvis, or lower urinary tract. In this case, SDS-PAGE detected low
molecular weight proteins within the urine and suggests that the primary lesion
is in the proximal tubules rather than the glomeruli.
8 This finding
is confirmed by the histopathology of the kidney in this case. The
characterization of proteinuria by SDS-PAGE is a useful antemortem clinical
tool to identify the main pathophysiologic mechanism involved.
Although not
reported in this case, this animal was likely in a secretory metabolic acidosis
due to renal tubular acidosis and loss of bicarbonate through the urine. In
cases of Fanconi-like syndrome, the renal proximal tubules fail to resorb
filtered bicarbonate.
3 Other
causes
of secretory metabolic acidosis include vomiting of intestinal contents rich in
bicarbonate, diarrhea, and an inability to swallow saliva rich in bicarbonate
in ruminants during dysphagia.3 The hallmark of this type of
acidosis is concurrent hyperchloremia as the body attempts to maintain
electroneutrality. In addition, the anion gap will typically be normal because
unmeasured anions are not increased.3
References:
1. Appleman EH,
Cianciolo R, Mosenco AS, Bounds ME, et al. Transient Acquired Fanconi Syndrome
Associated with Copper Storage Hepatopathy in 3 Dogs. J Vet Intern Med. 2008;
22:1038-1042.
2. Coronado VA,
ONeill B,Nanji M, Cox DW. Polymorphisms in canine ATP7B: candidate modifier of
copper toxicosis in the Bedlington terrier. Vet J. 2008; 2:293-96.
3. George JW,
Zabolotsky SM. Water, electrolytes, and acid base. In: Latimer KS, ed. Duncan
and Prasses Veterinary Laboratory Medicine Clinical Pathology. 5th
ed. Ames, IA: Iowa State University Press; 2011:163-166.
4. Hill TL,
Breitschwerdt EB, Cecere T, Vaden S. Concurrent Hepatic Copper Toxicosis and
Fanconis Syndrome in a Dog. J Vet Intern Med. 2008; 22:21922.
5. Hoffman G.
Copper-Associated Liver Diseases. Vet Clin Small Anim. 2009; 39:489511.
6. Hooper AN,
Roberts BK. Fanconi syndrome in four non-basenji dogs exposed to chicken jerky
treats. J Am Anim Hosp Assoc. 2011; 47:e178-87.
7. Langlois DK,
Smedley RC, Schall WD, Kruger JM. Acquired proximal renal tubular dysfunction
in 9 Labrador retrievers with copper-associated hepatitis (2006-2012). J Vet
Intern Med. 2013; 27:491-9.
8. Stockham SL,
Scott MA. Urinary system. In: Fundamentals of Veterinary Clinical Pathology. 2nd
ed. Ames, IA: Blackwell Publishing; 2008:458-460.
9. Thompson MF,
Fleeman LM, Kessell AE, Steenhard LA, Foster SF. Acquired proximal renal
tubulopathy in dogs exposed to a common dried chicken treat: retrospective
study of 108 cases (2007-2009). 2013. Aust Vet J. 9;368-73.