Signalment:
Ten-year-old,
mare, quarterhorse, (
Equus ferus caballus).This animal
presented to the teaching hospital in November of 2014 for weight loss. At that
time, blood work revealed hyperproteinemia and elevated blood ammonia (see
laboratory results below). Ultrasound of the liver revealed rounded margins. A
liver biopsy was performed and revealed nodular regeneration, bridging
fibrosis, and arteriolar and bile duct proliferation. The clinical suspicion at
that time was chronic pyrrolizidine alkaloid toxicity despite an absence of
hepatocellular cytomegaly or karyomegaly. The patient
re-presented in January of 2015 for increasing lethargy, dull mentation and
waxing and waning appetite. The horse was treated with minocycline,
metronidazole, pentoxifylline, lactulose, vitamin E, and prednisolone. The patient
presented again in February of 2015 due to progression of clinical signs and
failure to respond to treatment. Euthanasia was elected.
Gross Description:
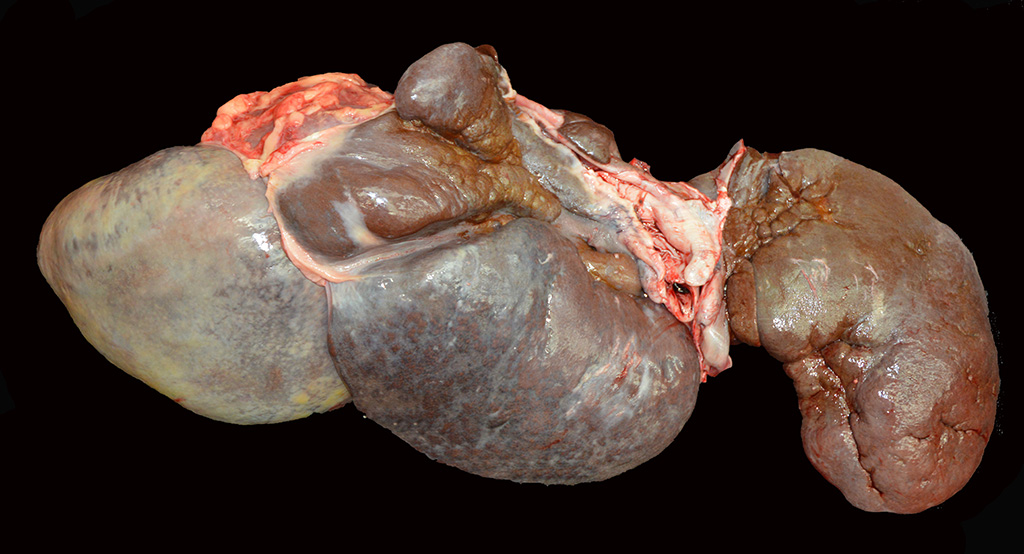
At
necropsy, the liver was markedly reduced in size weighing 3.1 kg (0.7% of body
weight) and had thick rounded margins. Some of the lobes appeared multinodular
and there were multifocal areas of capsular fibrosis.
Histopathologic Description:
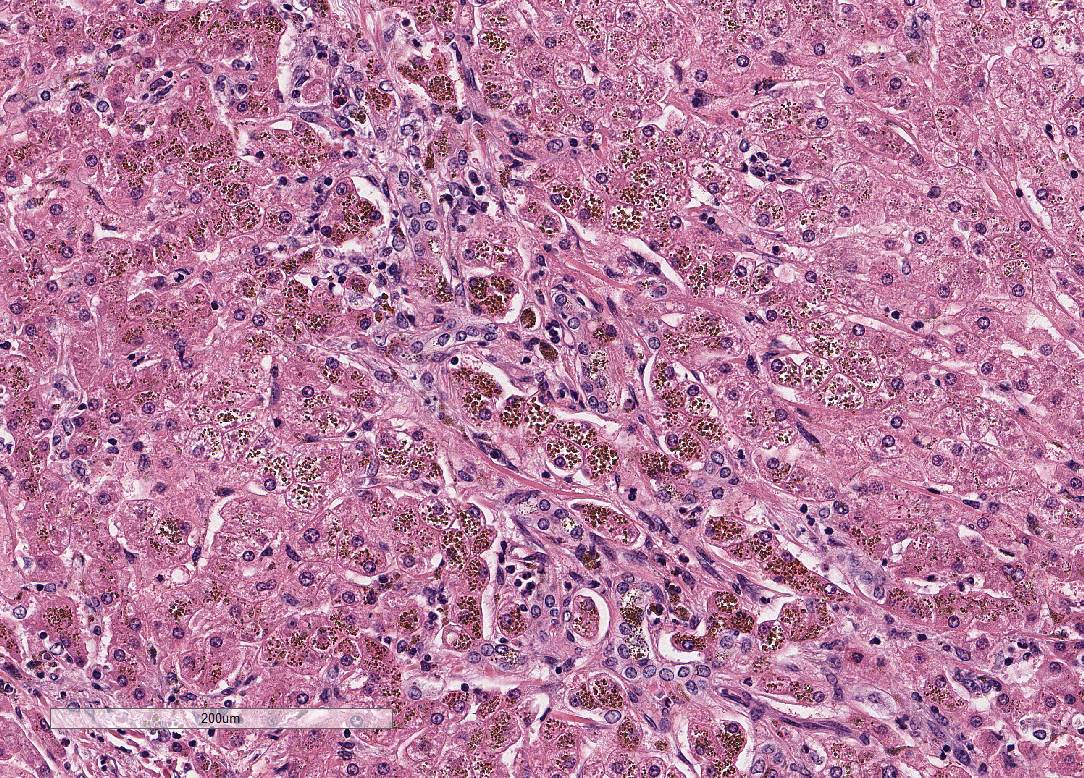
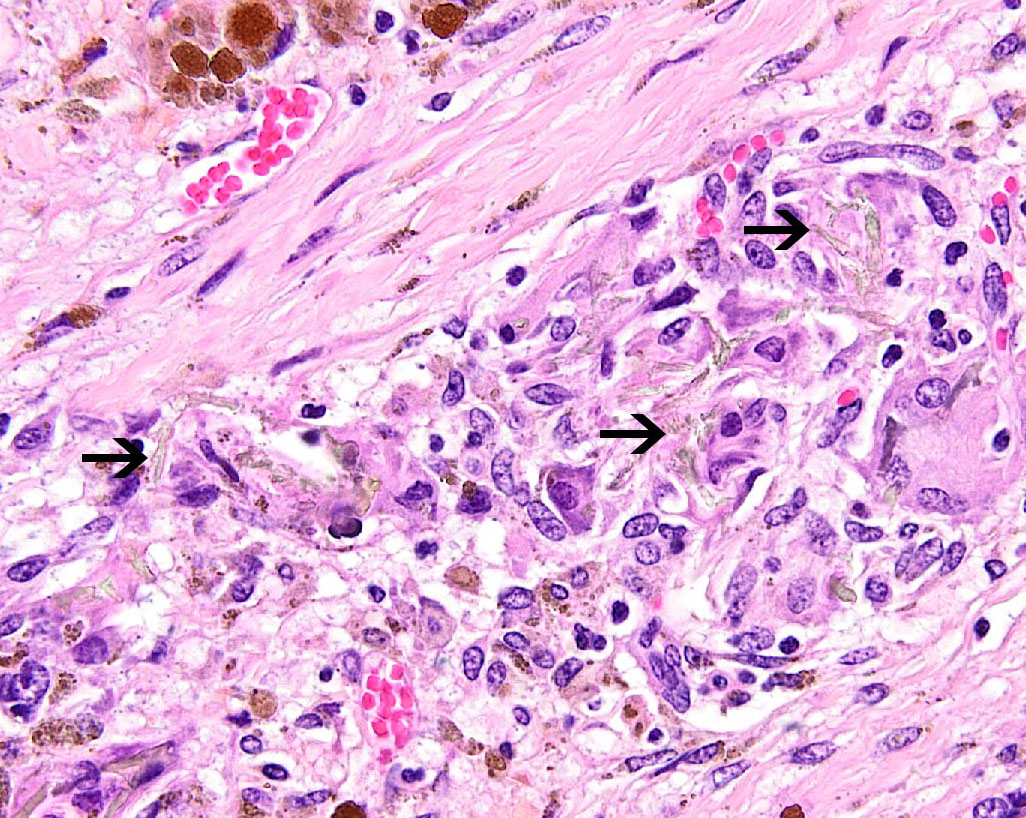
Liver:
Within all sections, the hepatic capsule is variably thickened, and the lobular
architecture is pronounced due to marked portal to portal bridging fibrosis.
Dissecting bands of fibrosis variably infiltrate the surrounding parenchyma
causing isolation of hepatic lobules and smaller clusters of hepatocytes. Rare
isolated hepatocytes appear hyper-eosinophilic with pyknotic to karyolytic
nuclei (individual cell apoptosis). Diffusely, hepatocytes contain variable
amounts of
coarse, dark
brown, granular pigment (hemosiderin). Kupffer cells and macrophages, which
course throughout areas of fibrosis, contain similar, but more variably sized,
dark brown cytoplasmic granules (hemosiderophages). Areas of fibrosis also
contain small to moderate numbers of lymphocytes and plasma cells, with rare
neutrophils, and abundant variably sized bile ducts and small-caliber blood
vessels. Occasional small aggregates of lymphocytes and plasma cells are
observed throughout the parenchyma. In some areas, the hepatic parenchyma has
been completely replaced by mature fibrous tissue and proliferating bile ducts.
In addition, mature fibrous tissue occasionally appears to occlude central and
portal veins with rare evidence of recanalization (venoocclusive disease).
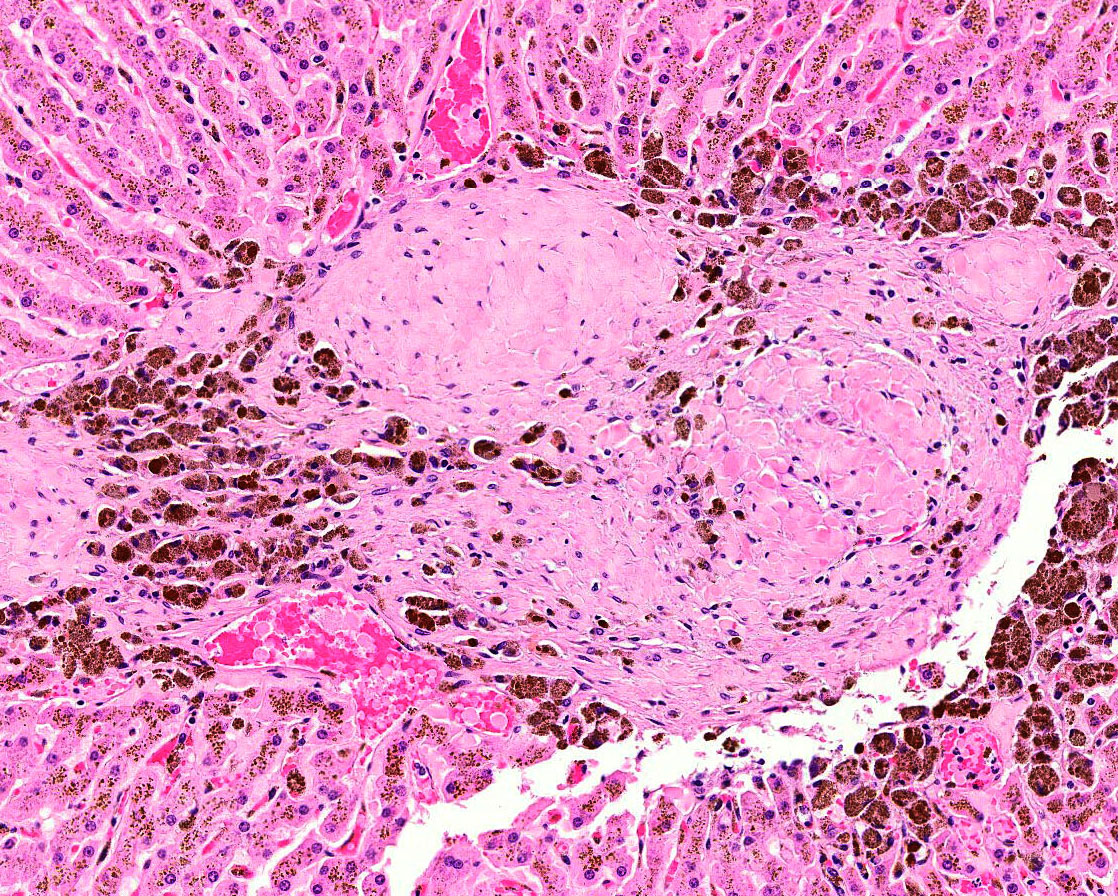
Within one section there are large, multifocal areas of acute hemorrhage and
edema that cause separation and dis-organization of the hepatic cords.
Occasional individual collagen bundles are segmentally darkly amphophilic
(sidero-calcinosis). Within the most severely affected sections, there are
frequent, irregular, refractile crystals associated with small numbers of
multinucleated giant cells and epithelioid macrophages (foreign body response).
Morphologic Diagnosis:
Liver: Severe, chronic, bridging portal and capsular fibrosis with lobular
collapse, veno-occlusion, biliary hyperplasia and marked hepatocellular
hemosiderosis.
Lab Results:
February 2015:
(reference ranges)
GGT 294 IU/L
(8-22)
SDH 16 IU/L
(0-8)
Resting bile
acids 36 uMOL/L (4-11.5)
Plasma ammonia
242 UG/DL (5-59)
Postmortem liver
heavy metal screen
Iron 5100 ppm
(100-300)
Condition:
Haemochromatosis
Contributor Comment:
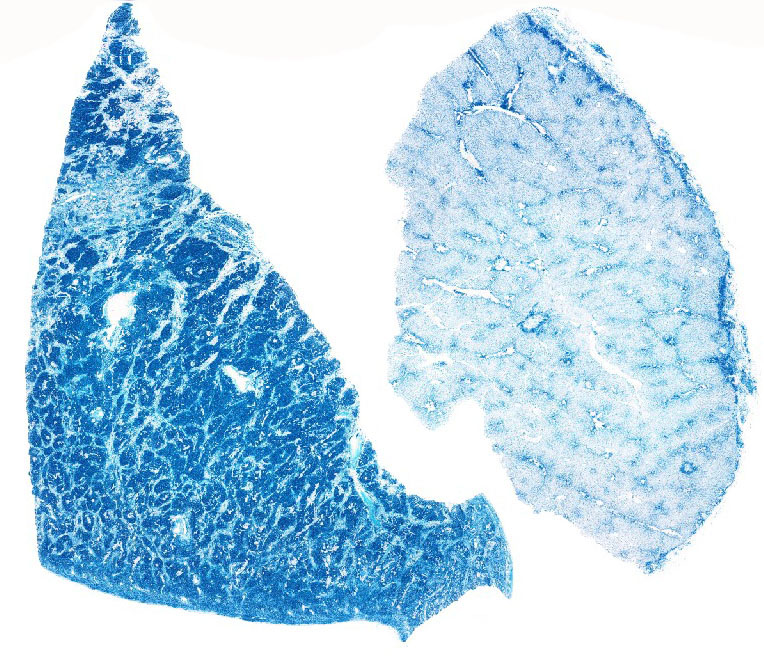
The abundant hepatocellular and Kupffer cell pigment was confirmed as iron
using Perls iron stain. Heavy metal analysis of the liver revealed an iron
concentration of 5100 ppm, far exceeding the normal upper limit of 300 ppm.
Perls staining also revealed iron deposits within smooth muscle trabeculae of
the spleen, and within renal tubular epithelium at the corticomedullary
junction. Iron within splenic trabeculae and within fibrous tissue in the liver
is occasionally deposited in conjunction with calcium salts (siderocalcinosis).
In addition, the most severely affected sections shows deposition of refractile
clear crystals that are associated with a granulomatous foreign body response.
These crystals are not birefringent under polarized light.
With the
exception of certain avian species, including mynahs and toucans, hemo-chromatosis
is a rare condition in domestic species. A hereditary form of hemochromatosis
has been reported in Salers and Salers-cross cattle,
8 and there have
been individual case reports in horses.
6 Cases of dietary iron
overload have also been reported in sheep and cattle. Iron storage disease is
classically divided into two entities: hemosiderosis (iron overload in the
absence of clinical signs) and hemochromatosis (iron overload leading to
hepatic damage including fibrosis, inflammation, and liver failure). In human
medicine, iron storage disease is additionally subdivided into primary and
secondary categories. Primary iron storage disease involves an inherent
abnormality in iron metabolism. In humans, this is most commonly the result of
one of two missense mutations in the HFE gene which encodes a protein involved
in the interaction between transferrin and the transferrin receptor.
9
Secondary hemochromatosis occurs from excessive intestinal absorption of iron
either due to iron excess within the diet, or as a response to increased demand
for erythro-poiesis. Secondary hemochromatosis can occur with any condition
leading to chronic hemolysis. Although a very small amount of iron is
eliminated through the bile, the body has no natural way of responding to
excess iron and therefore iron accumulates over time, primarily in the liver.
Iron accumulation results in hepatocellular toxicity through production of free
radicals and organelle dysfunction, including lysosomal injury.
8
This can lead to hepatocellular necrosis that progresses to bridging fibrosis,
bile duct hyperplasia, and venoocclusive disease, as observed in this case. In
humans, hemochromatosis can also increase the risk of hepatocellular neoplasia
9
and in birds, has been shown to predispose to certain bacterial infections such
as
Yersinia pseudotuberculosis.
4
Distinguishing
between primary and secondary iron storage disease can be especially difficult
in chronic cases. In humans, the pattern of hemosiderin deposition can be
helpful.
6 In primary iron storage disease, hemosiderin accumulates
first in hepatocytes while in secondary hemosiderosis, iron accumulates first
within Kupffer cells and macrophages (reticulo-endothelial system). However,
this requires that the liver is examined early in disease progression. In this
case, a primary abnormality in iron metabolism is suspected based on the lack
of clinical or histologic evidence of a second underlying disease process
leading to chronic hemolysis, and feeding of a standard equine diet.
JPC Diagnosis:
Liver:
Fibrosis, portal and bridging, diffuse, marked with hepatocellular degeneration
and loss and intra-hepatocellular hemo-siderosis, Quarterhorse,
Equus ferus
caballus.
Conference Comment:
The contributor provides an outstanding example and thorough review of
hemochromatosis in humans and veterinary species. Free iron is highly toxic to
tissues due to its ability to participate in the generation of hydroxyl radical
formation via the Fenton or Haber-Weiss reaction with hydrogen peroxide (H
2O
2)
leading to lipid peroxidation and DNA damage.
5 As a result, iron is
typically bound to transferrin while in circulation and either ferritin or
hemosiderin when stored and sequestered in tissue. When iron is bound to these
proteins, it cannot participate in these injurious reactions. Ferritin
concentration is highest in the liver, spleen, and bone marrow and is stored in
hepatocytes and/or macrophages.
1,3,5,7
In hepatocytes, iron is derived from plasma transferrin,
while iron stored within macrophages is a result of erythrocyte breakdown.
Normally, to offset the attritional loss of daily iron, duodenal enterocytes
absorb approximately 1 to 2 mg of iron per day from the diet via divalent metal
transporter-1 (DMT-1) and a heme carrier protein-1 (HCP-1) on the luminal
surface of the enterocytes.
5,9 Iron is transported from the
cytoplasm of the enterocyte to the circulation by ferroportin.
1,7
Absorbed iron circulates bound to transferrin and is used primarily by
erythroid precursors in the synthesis of heme. Macrophages in the spleen clear
dead and dying erythrocytes and release the iron from heme to export it to the
circulation or store it in ferritin.
In
iron-overload, transferrin is quickly saturated and iron is stored in the liver
and various other tissues due to the lack of a regulated pathway for effective
iron excretion.
3
As mentioned above, hepatocytes are a major site of iron
storage as ferritin and are also responsible for the production of type II
acute phase protein, hepcidin, in response to inflammatory cytokine,
interleukin-6.
1,3,5,7 Hepcidin is transported in by blood by alpha-2-macroglubulin
and blocks the release of iron from enterocytes and macrophages by degrading
the iron exporter, ferroportin. In humans, decreased hepcidin synthesis caused
by mutations in the hepcidin gene,
HAMP, causes severe hemochromatosis
in juveniles.
1,5
Within the cytoplasm, iron is stored as ferritin, which is
reconverted into iron as needed by the body. If tissue ferritin levels are
high, ferritin aggregates into hemo-siderin globules, which is much more
difficult to revert back to free iron. In hepatocytes overwhelmed with iron,
most iron is stored as hemosiderin. Ferritin and hemosiderin readily stain with
Pearls Prussian blue, demonstrated nicely in this case.
1,5
Iron is a direct hepatotoxin and iron overload often results
in the formation periportal bridging fibrosis with little inflammation.
1,5
In addition to the markedly elevated postmortem liver heavy metal screen (Iron:
5100 ppm [100-300]), clinical pathology data from the provided serum
biochemistry supports the histologic findings in this case. Elevated sorbital
dehydrogenase (SHD: 16 IU/L [0-8]) and elevated plasma ammonia (242 UG/dL
[5-59]) indicate hepatocellular injury and decreased hepatic function
respectively. In addition, elevated gamma-glutamyl trans-peptidase (GGT: 294
IU/L [8-22]) and elevated resting bile acids (36 uMOL/L [0-20]) indicate
cholestasis and biliary hyperplasia with decreased hepatobiliary function.
1,2,5,8
References:
1. Bain PJ. Liver.
In: Latimer KS ed.
Duncan and Prasses Veterinary Laboratory Medicine
Clinical Pathology. 5
th ed. Ames, IA:Wiley-Blackwell;
2011:213-224.
2. Brockus CW.
Erythrocytes. In: Latimer KS ed.
Duncan and Prasses Veterinary Laboratory
Medicine Clinical Pathology. 5
th ed. Ames, IA:Wiley-Blackwell;
2011:4
3. Cullen JM,
Stalker MJ. Liver and biliary system. In: Maxie MG, ed.
Jubb, Kennedy, and
Palmers Pathology of Domestic Animals. Vol 2. 6th ed. Philadelphia,
PA:Elsevier; 2016:272.
4. Galosi L,
Farneti S, Rossi G, et al
. Yersinia pseudotuberculosis, serogroup O:1A,
infection in two amazon parrots (
Amazona aestiva and
Amazona oratrix)
with hepatic hemosiderosis.
J Zoo Wildl Med. 2015; 46(3):588-591.
5. Kumar V, Abbas
AK, Fausto N. Red blood cell and bleeding disorders. In:
Robbins and Cotran
Pathologic Basis of Disease. 9th ed. Philadelphia, PA:Elsevier Saunders;
2015:650-651.
6. Lavoie JP,
Tuescher JP. Massive iron overload and liver fibrosis resembling
haemochromatosis in a racing pony.
Equine vet J.
1993;25(6):552-554
7. Mazzaro LM,
Johnson SP, Fair PA, Bossart G, Carlin KP, Jensen ED, Smith CR, Andrews GA,
Chavey PS, Venn-Watson S. Iron indices in bottlenose dolphins (Tursiops
truncatus).
Comp Med. 2012; 62:508-515.
8. OToole D, Kelly
EJ, McAllister MM, et al. Hepatic failure and hemochromatosis of Salers and
Salers-cross cattle.
Vet Pathol. 2001; 38(4):372-389.
9. Pietrangelo A.
Hereditary hemochromatosisA new look at an old disease.
N Engl J Med.
2004; 350(23):2383-2397.
10. Weiss DJ. Iron
and copper deficiencies and disorders of iron metabolism. In: Weiss DJ, Wardrop
KJ, eds.
Schalms Veterinary Hematology. 6
th ed. Ames,
IA:Wiley-Blackwell; 2010:170.