Signalment:
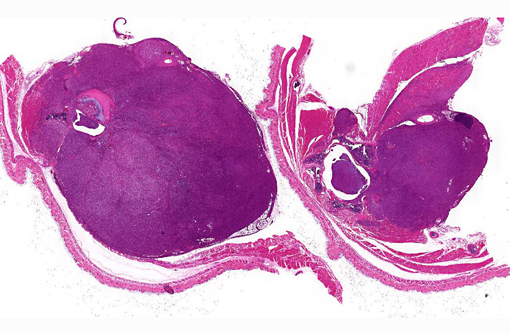
Peritoneal cavity: Lumbosacral soft tissue mass.
Histopathologic Description:
Morphologic Diagnosis:
Spinal cord: Compressive unilateral leukomalacic myelitis, white matter degeneration and regeneration, and spinal ganglia neuritis, chronic-active.
Condition:
Contributor Comment:
The most effective model for characterizing the structure and function of dystrophin and possible therapeutic interventions for DMD is the mdx mouse.(1) The official nomenclature of mdx mice is C57BL/10Scsn-Dmdmdx/J. A point mutation in exon 23 of the x-linked dystrophin gene (dmd) creates a nonsense mutation that converts cytosine to thymine. This substitution replaces a glutamine codon with a termination codon, causing abnormal production, and/or reduced stability of truncated gene products. In mdx mice, skeletal muscle has normal histologic features until about 3 weeks of age.(4) It then undergoes progressive degeneration and necrosis; small caliber fibers with central nuclei can be observed as part of the regenerative response.(4) In mice, the mutated dystrophin gene does not manifest with severe muscular dystrophy, as it does in humans, due to compensatory responses by utrophin. It does however have a somewhat shortened lifespan,(1) though not as dramatic as in humans. In mice with both utrophin and dystrophin knocked out, there is more severe disease and premature death.Â
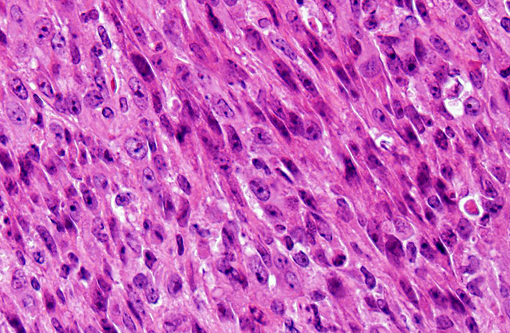
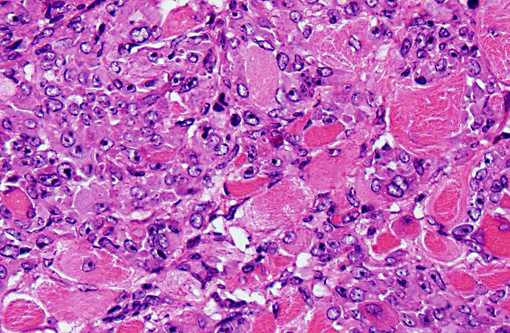
In our facility and others,(1) mdx mice show a tendency toward developing spontaneous rhabodmyosarcomas. They can occur on the distal limb or the trunk, as in this case. There is no limb predilection.(1) Rhabdomyosarcoma is a malignant tumor of striated muscle that is, in veterinary medicine, divided into four major histologic categories: embryonal, botryoid, alveolar, and pleiomorphic {Mueuten, 2002 #129}. Diagnostic features of rhabdomyosarcoma include elongate strap cells, racket cells, as well as cross striations which can be highlighted with phosphotungstic acid-hematoxylin stain (PTAH).(5) These tumors can also be labeled with myosin, actin, desmin, vimentin, BB creatine kinase, NCAM, IFG-II and TGF-Beta.(5) Rhabdomyosarcoma in mice has been shown to express the myogenic differentiation factors myogenin, MyoD, and the muscle intermediate filament protein desmin.(3) It is speculated that mdx mice are predisposed because of the lifelong continuous myofiber degeneration and regeneration, which is associated with continuous and massive activation and proliferation of satellite cells (muscle progenitor cells), increasing the chance of developing random and spontaneous mutations.(2) Inactivation of p53 is a primary event in mdx rhabodmyosarcoma.(3)
Other animal models of DMD include dogs, cats, zebrafish, and C. elegans.(2) In dogs, there is an X-linked muscular dystrophy in several breeds, including golden retrievers, Rottweilers, German short-haired pointers, and beagles. The manifestation in golden retriever is most closely homologous model of DMD. In this breed, the disease results from a single base pair change in the 3 consensus splice site of intron 6, which leads to skipping of exon 7 and a misaligned reading frame in exon 8 that causes a premature stop codon.(11) The myocardium is more severely affected in the golden retriever than in other animal models, though this feature of the disease course makes it much closer to the manifestation in humans.(11) Cats have a hypertrophic feline muscular dystrophy that has limited similarity to DMD. In cats, the disease is due to a 200 kb deletion of the dystrophin gene, which causes a hypertrophic muscular dystrophy.(11) Affected cats typically have elevated creatine kinase in the blood by 4-5 weeks of age, before apparent muscle involvement, which can be seen at 10-14 weeks.(11) Affected cats die of esophageal compression by a hypertrophic diaphragm, or of the inability to drink due to glossal hypertrophy.(11) Zebrafish and C. elegans express a dystrophin homologue that is used for gene analysis and drug discovery. There are no primate models of DMD.(11)
JPC Diagnosis:
1. Vertebral body and epaxial musculature: Rhabdomyosarcoma.
2. Spinal cord: Leukomalacia, focally extensive, moderate.
Conference Comment:
Table 1: Animal models of muscular dystrophy.2,6,7-9,11
| Model | Species | Defect | Other |
| X-linked muscular dystrophy (mdx; Duchennes-like) | mdx mouse | X-linked dystrophin defect | No muscle wasting due to compensatory responses by utrophin |
| Human classical congenital muscular dystrophy | dy+/dy+ mouse | Autosomal recessive laminin alpha 2 (merosin) deficient | Loss of myelin in ventral nerve roots |
| X-linked muscular dystrophy (xmd; Duchennes-like) | Dog | X-linked dystrophin defect | Best characterized in golden retriever; myocardium more severely affected than other muscles |
| Hypertrophic feline muscular dystrophy | Cat | X-linked dystrophin defect | Protruding tongue, bunny-hopping gait; malignant hyperthermia-like syndrome |
| Hereditary muscular dystrophy | Chicken | Autosomal dominant defect in Ubiquitin ligase gene (WWP1) | Superficial pectoralis (large breast muscle); affects type II muscle fibers |
| Ovine muscular dystrophy | Merino sheep | Autosomal recessive | Australia |
| Muscular dystrophy | Meuse-Rhine-Yssl cattle (Netherlands); rarely in Holstein-Friesians | Probably autosomal recessive | Usually affects diaphragm |
References:
2. Collins CA, Morgan JE. Duchenne's muscular dystrophy: animal models used to investigate pathogenesis and develop therapeutic strategies. Int J Exp Pathol. 2003;84:165-172.
3. Fernandez K, Serinagaoglu Y, Hammond S, Martin LT, Martin PT. Mice lacking dystrophin or alpha sarcoglycan spontaneously develop embryonal rhabdomyosarcoma with cancer-associated p53 mutations and alternatively spliced or mutant Mdm2 transcripts. Am J Pathol. 2010;176:416-434.
4. The Jackson Laboratory, JAX mice database- C57BL/10ScSn-Dmdmdx/J. http://jaxmice.jax.org/strain/001801.html.
5. Maronpot RR. Pathology of the Mouse: Reference and Atlas. Vienna, IL: Cache River Press; 1999:637-642.
6. Matsumoto H, Maruse H, Inaba Y, et al. The ubiquitin ligase gene (WWP1) is responsible for the chicken muscular dystrophy. FEBS lett. 2008;582(15): 2212-2218.
7. Nakamura N. Dystrophy of the diaphragmatic muscles in Holstein-Friesian steers. J Vet Med Sci. 1996;58(1): 79-80.Â
8. van Lunteren E, Moyer M, Leahy P. Gene expression profiling of diaphragm muscle in alpha2-laminin (merosin)-deficient dy/dy dystrophic mice. Physiol Genomics. 2006;25(1): 85-95.
9. van Vleet JF, Valentine BA. Muscle and tendon. In: Maxie MG, ed. Jubb, Kennedy, and Palmers Pathology of Domestic Animals. Vol 1. 5th ed. Philadelphia, PA: Elsevier Limited; 2007:210-216.
10. Wang Z, Chamberlain JS, Tapscott SJ, Storb R. Gene therapy in large animal models of muscular dystrophy. ILAR J. 2009;50:187-198.
11. Willmann R, Possekel S, Dubach-Powell J, Meier T, Ruegg MA. Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul Disord. 2009;19: 241-249.