Signalment:
Gross Description:
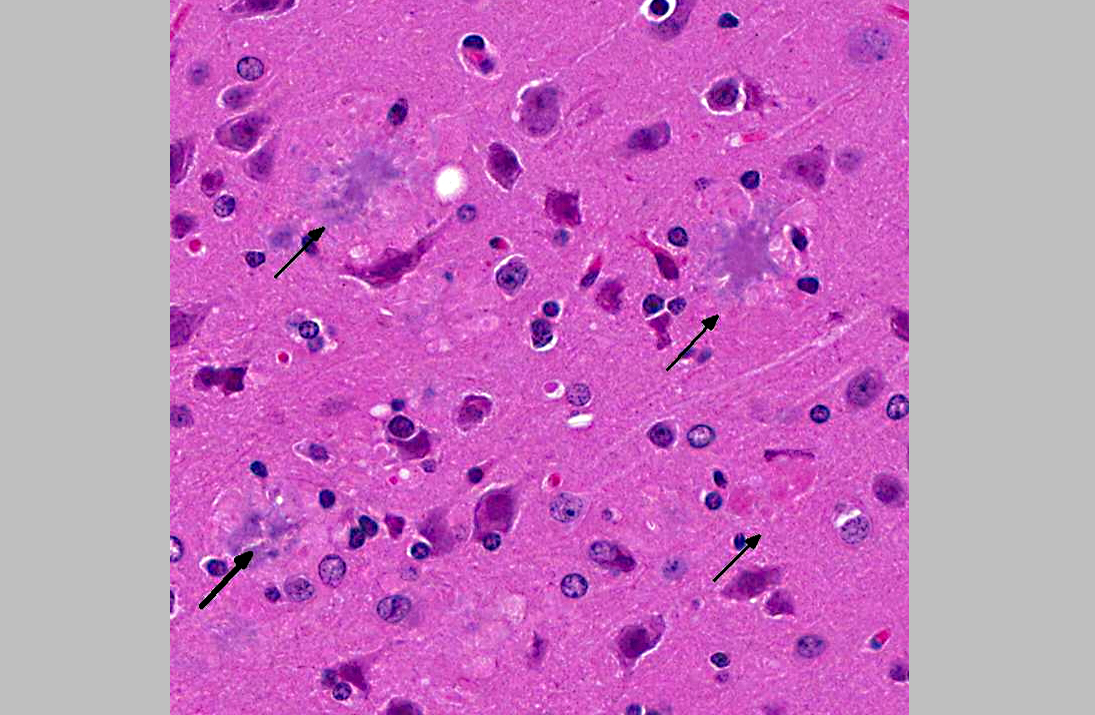
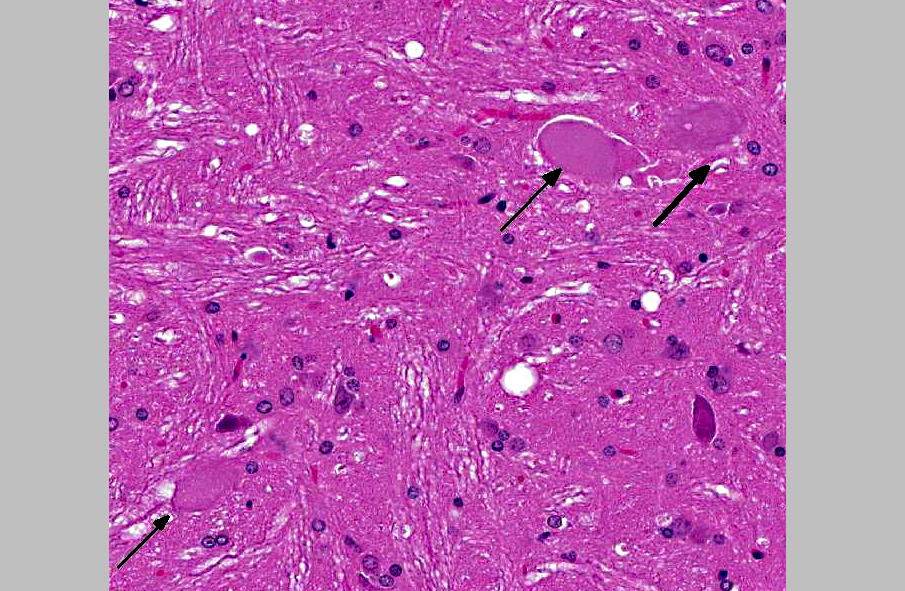
Histopathologic Description:
Morphologic Diagnosis:
Condition:
Contributor Comment:
The main component of the amyloid core is A, a peptide derived from cleavage of the larger protein Amyloid Precursor Protein (APP), a cell surface protein that may function as a receptor.(2) The A peptides readily aggregate and can be directly neurotoxic and can result in synaptic dysfunction and an inflammatory response. Other proteins that present in plaques in smaller amounts include complement proteins, pro-inflammatory cytokines, 1 -antichymotrypsin, and apolipoproteins. Findings in individuals affected by the familiar form of AD have supported the hypothesis that the generation of A is a critical step in the initiation of the disease. Some patients with familial AD have point mutations of the APP gene. Two other commonly affected loci are in the Presenilin 1 and 2 (PS1, PS2), which lead to a gain of function of the -secretase complex, which is involved in the cleavage of APP into A.
The other main morphologic change in AD is the formation of neurofibrillary tangles, which are composed of bundles of filaments in the cytoplasm of neurons.(2) These are seen as basophilic structures by H&E staining, and can be demonstrated by silver stain methods such as Bielschowsky. A major component of these filaments is a hyperphosphorylated form of the protein tau. Tangles are not specific to AD, as they can be found in other diseases. There was no evidence of neurofibrillary tangles in the brain of these mice on H&E and Bielschowsky stains.
JPC Diagnosis:
1. Cerebrum, hippocampus, amygdaloid nucleus and brainstem: Neuritic plaques, numerous, diffuse, with gliosis and neuronal loss.
2. Brainstem nuclei and neurons: Chromatolysis, multifocal, marked, with spheroid formation.
Conference Comment:
Three autophagic mechanisms are described in mammalian cells: chaperone-mediated autophagy, microautophagy, and macroautophagy, all which result in the lysosomal degradation of targeted cellular components. In chaperone-mediated autophagy, cytoplasmic proteins exposing a KFERQ-like motif are targeted directly to the lysosomes for degradation. Microautophagy involves the invagination of the lysosomal membrane to nonselectively engulf and degrade smal l por t ions of the cytoplasm. Macroautophagy is the best characterized mechanism of autophagy; thus, it is often referred to as simply autophagy. Unlike chaperone-mediated autophagy and microautophagy, macroautophagy relies on the de novo synthesis of double membrane-bound autophagosomes. The major upstream inhibitor of macroautophagy initiation is the mammalian target of rapamycin complex 1 (mTORC1), which regulates several cellular processes, to include autophagy, cell growth and proliferation, and protein synthesis. Various cell signaling pathways converge upon mTORC1; for instance, in response to insulin and growth factors, the class I phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway activates mTORC1 and thus suppresses autophagy. Conversely, energy depletion signals through the liver kinase B1 (LKB1)/AMPactivated protein kinase (AMPK) pathway to inhibit mTORC1, and thereby activate autophagy.
When mTORC1 is inhibited, autophagy-related (Atg) proteins coordinate a series of events that results in the formation of an autophagosome (i.e., vesicle nucleation, vesicle elongation, docking and fusion, and vesicle maturation). This series begins with the formation of a phagophore (i.e., pre-autophagosomal structure) and progresses to the formation of a double membrane enclosed autophagosome, the outer membrane of which fuses with a lysosome or a late endosome to form an autolysosome or an amphisome, respectively. Acidic hydrolases digest material within the autolysosome or amphisome, after which the lysosome is restored.(5)
Current research strongly suggests the disruption of proteolysis within autolysosomes is the principle mechanism underlying autophagy failure in Alzheimers disease. Various forms of autophagic vacuoles representing intermediate stages in autophagy (including autophagosomes, amphisomes, and autolysosomes) have been observed to accumulate in the brains of patients with Alzheimers disease and other neurodegenerative diseases.(4,5) Interestingly, attempts to restore lysosomal proteolysis and enhance autophagy in mouse models of Alzheimers disease have shown positive effects on neuronal function and cognitive performance.(5)
References:
2. Oakley H, Cole SL, Logan S, et al. Intraneuronal amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimers disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129-10140.
3. Gibbs G. Alois Alzheimer: The Man. http://www.unmc .edu/intmed/geriatrics/docs/alois_alzheimer.pdf Accessed 19 October 2012.
4. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27-42.
5. Nixon RA, Yang DS. Autophagy failure in Alzheimers disease-�-�Locating the Primary Defect. Neurobiol Dis. 2011; 43(1):38-45.