Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2018-2019
Conference 3
Sept 19, 2018
CASE III: P2551-13 (JPC 4048996-00).
Signalment: 3 year-old, female, Standardbred, Equus caballus, horse
History: This mare had a history of decreased racing performances of a few weeks duration. A week prior to its last race, an endoscopic examination was performed and revealed excessive tracheal mucus; it was treated with enrofloxacin for a week. The morning after its last race, the horse was anorectic and listless/depressed; it was treated with IV trimethoprim-sulfa, DMSO and electrolytes. The condition rapidly worsened over the next 24 hours, with tachypnea, marked weakness and fever. Clinical biochemistry revealed hypercreatininemia (800 µmol/L; normal range: 87-150). Tracheobronchial washing results were within normal limits. Acute renal failure was strongly suspected. Due to the poor prognosis, the owners elected for euthanasia and a full necropsy was performed.
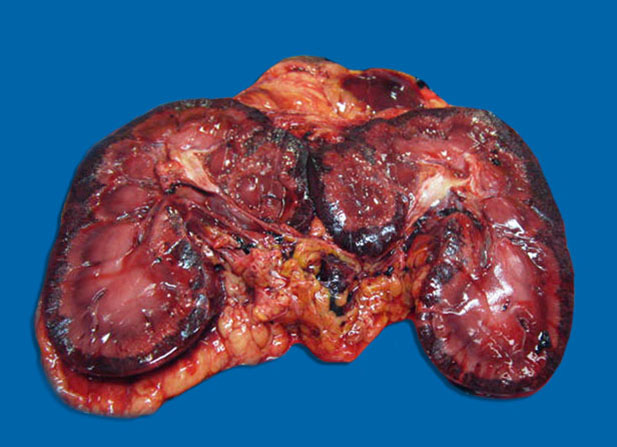
Gross Pathology: There was subcutaneous edema with multifocal petechiation in the dependent portions of the abdomen. Approximately 1.5 L of blood-tinged serous fluid was present in the thoracic cavity. The lungs showed moderate diffuse congestion and edema with a few small (< 1 cm) randomly distributed dark red foci. The liver was pale and appeared slightly yellowish. The cortex of both kidneys was diffusely hemorrhagic. Petechiae were present multifocally on the small intestinal mucosa, sometimes accompanied by mild submucosal edema. Colonic contents were slightly more fluid than normal, without any associated lesions.
Laboratory results: Routine aerobic bacteriology on ileum and colon yielded 4+ of an α-hemolytic Streptococcus (not further identified) and 1+ of E.coli (negative for Salmonella). The latter was negative by PCR for all tested virulence factors including Stx1 and Stx2.
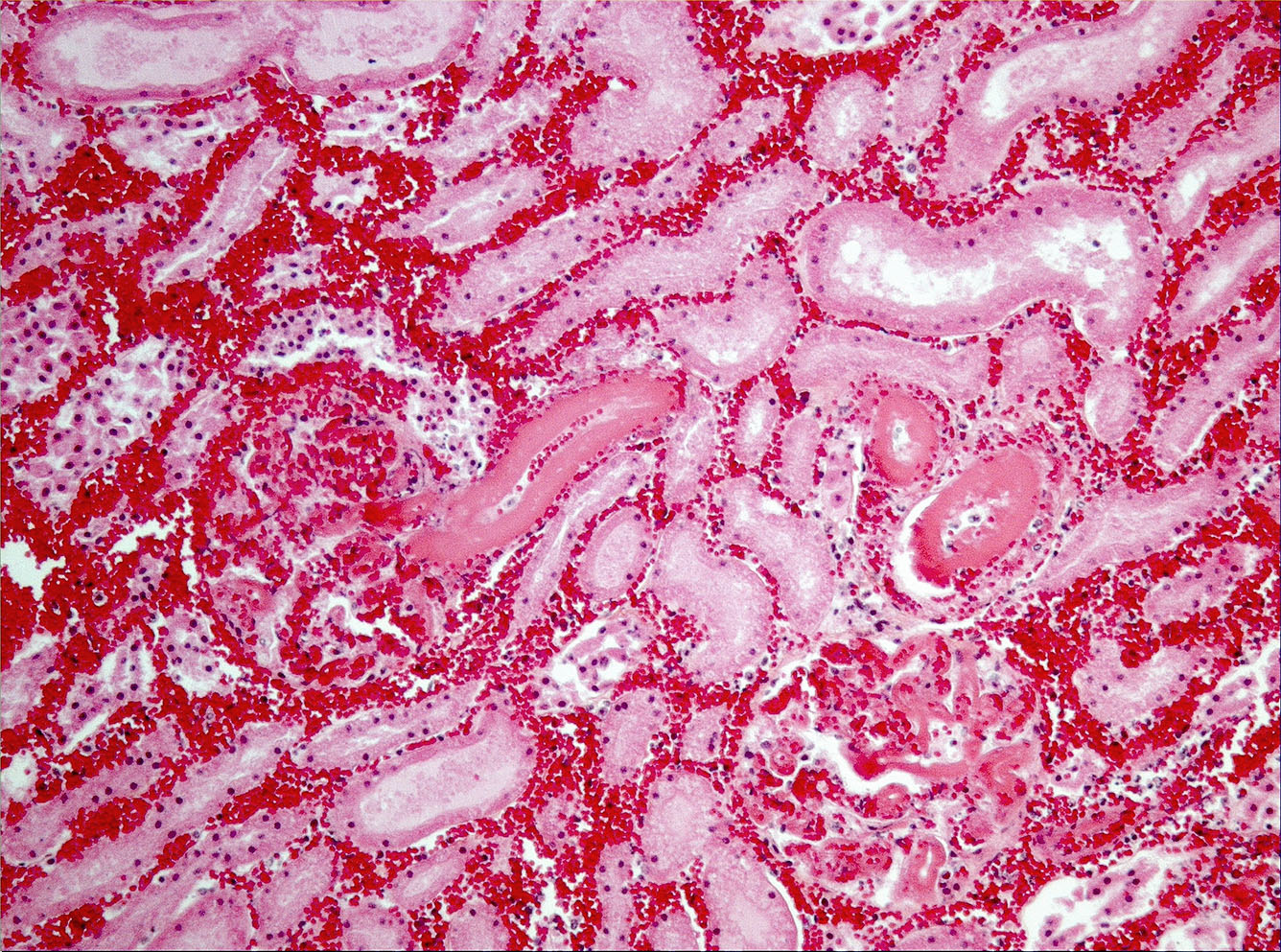
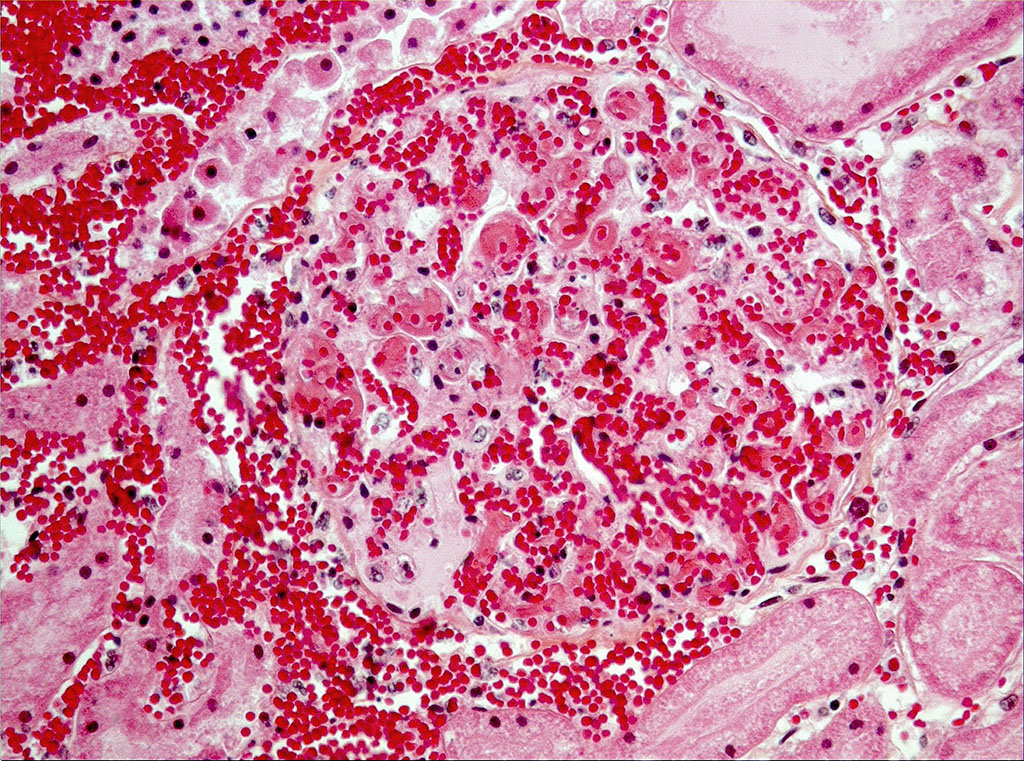
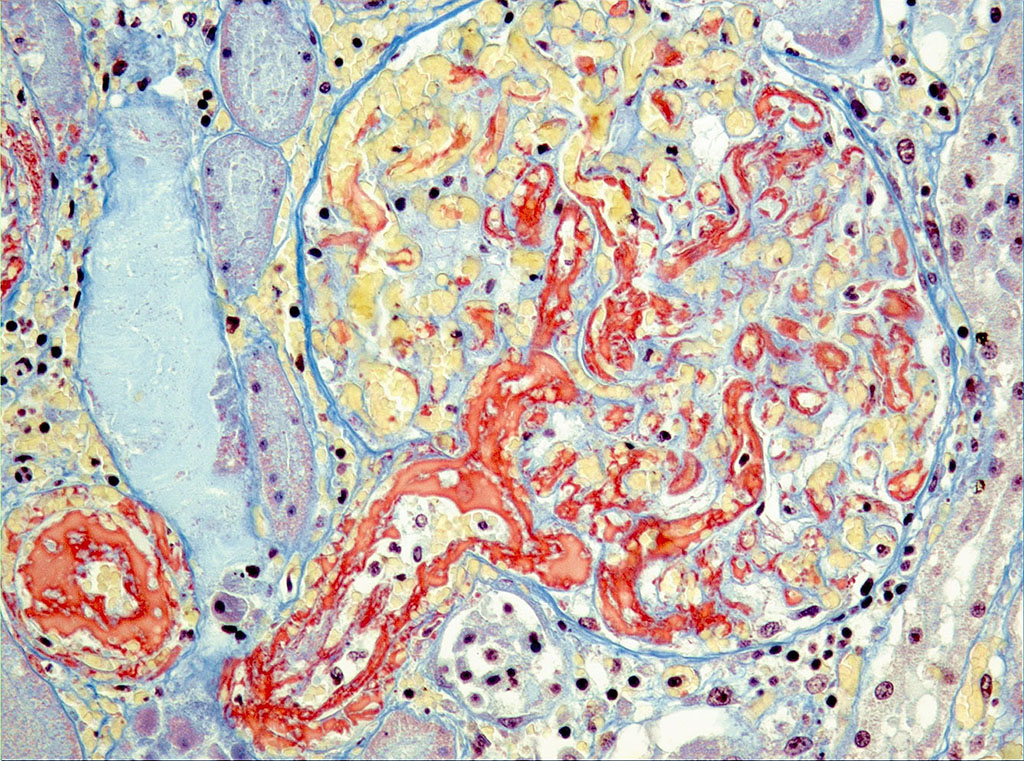
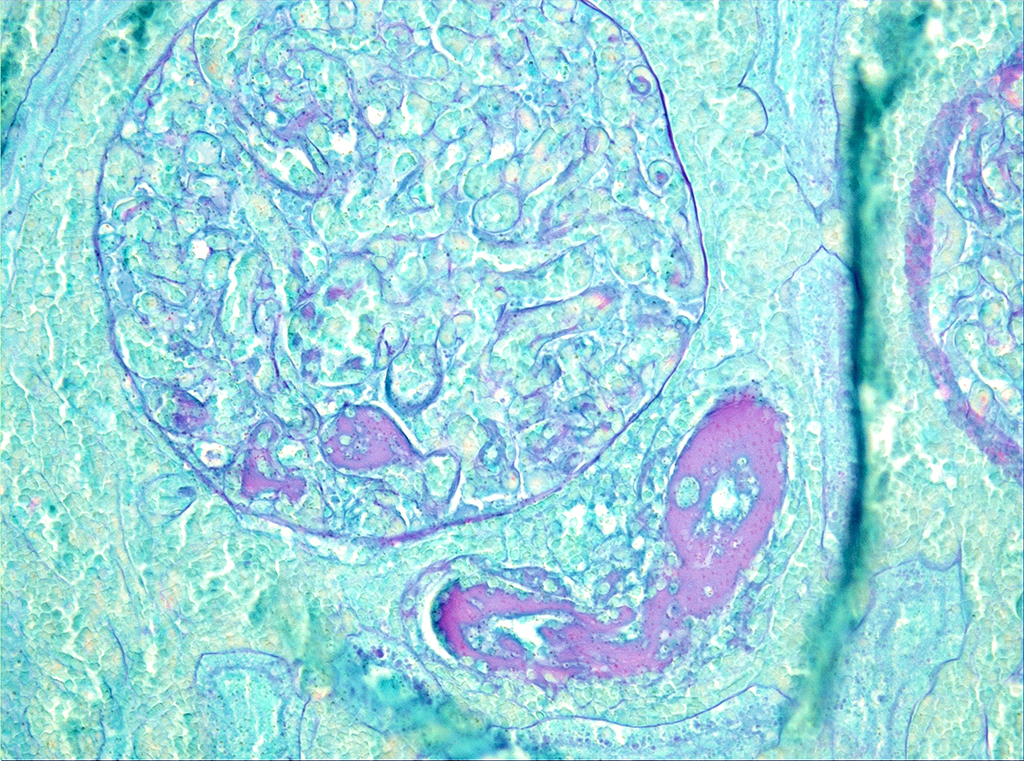
Microscopic Description: On low magnification, the renal cortex has a variegated appearance. There are severe and extensive cortical interstitial hemorrhages associated with vascular lesions involving glomerular capillaries, interlobular and (afferent) glomerular arterioles, and fewer venules. These lesions consist of hyaline thrombi and thickening of vascular walls by fibrinoid material with nuclear debris (fibrinoid necrosis). The fibrinoid material is often peripheral, luminal and parietal (the limit is often blurred), and associated with erythrocytes; the fibrinous nature is supported by its Martius scarlet blue positivity. The fibrinoid vascular material is PAS-positive and Jones’ silver-negative. Especially in non-hemorrhagic areas, the capillaries in some glomeruli contain granular material (possible platelets), fragmented erythrocytes and/or thrombi. There is multifocal acute tubular necrosis in the cortex and superficial medulla, the extent of which is hard to evaluate (some post-mortem change and very acute), and tubular proteinosis. Inflammation is minimal: occasionally a few interstitial degenerate neutrophils and small lymphocytes.
Contributor’s Morphologic Diagnoses: Severe, extensive renal necrotizing vasculopathy with fibrin thrombi (glomerular capillaries, interlobular and afferent glomerular arterioles) and extensive hemorrhage (consistent with hemolytic-uremic syndrome).
Multifocal, acute tubular necrosis (ischemic, secondary to vascular lesions).
Contributor’s Comment: Even though hematological data was not available (i.e. to assess microangiopathic hemolytic anemia and thrombocytopenia), hemolytic-uremic syndrome was the final diagnosis, based on the renal lesions associated with acute renal failure. Unfortunately, no cause was found, but only Shiga toxin-producing E. coli (STEC) were investigated.
Hemolytic-uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) are the two best-known thrombotic microangiopathies, which are clinical syndromes characterized by microangiopathic hemolytic anemia, thrombocytopenia and dysfunction of one or more organs, the result of microvascular thrombosis and endothelial damage. Both HUS and TTP cause the formation of platelet-rich thrombi in the microcirculation and endothelial swelling, which cause erythrocyte fragmentation (leading to anemia) and excessive platelet consumption (leading to thrombocytopenia).1,7,10 In their original description, HUS involved mostly or only the kidney, with acute renal failure as a prominent feature, while TTP involved mostly the brain, with mainly neurologic manifestations. However, it is now known that HUS can involve organs other than the kidney, including the brain (and thus cause neurologic dysfunction), and TTP patients often have renal involvement; thus, these two clinical syndromes often overlap.7,10 Thrombotic microangiopathies in humans are now classified, based on known causes/pathogenesis or associations, as 1) typical HUS (epidemic, classic or diarrhea-positive HUS), atypical HUS (non-epidemic or diarrhea-negative HUS) and TTP. Typical HUS (tHUS) is caused by intestinal Shiga toxin-producing bacteria, in most cases E.coli producing Stx1 ot Stx2 (STEC), and occasionally other bacteria like Shigella dysenteriae type 1; the best known STEC serotype is the O157:H7, which is most often acquired by ingestion of undercooked contaminated ground beef (thus its sometimes epidemic nature). Atypical HUS (aHUS) is often associated with a genetic (mutation) or, less commonly, acquired (autoantibodies) deficiency of complement regulatory proteins, mostly factor H (CFH), predisposing to hyperactivation of the alternative complement pathway. Thrombotic thrombocytopenic purpura (TTP) is associated with an acquired (autoantibodies) or, less commonly, a genetic (mutation) deficiency in ADAMTS13, a metalloproteinase which degrades high-molecular-weight multimers of von Willebrand Factor (vWF); subsequent accumulation of ultralarge vWFmultimers (UL-VWF) in plasma promotes platelet aggregation and activation. Triggering conditions or exposures associated with aHUS and TTP include pregnancy, autoimmune diseases and certain drugs (e.g. cyclophosphamide).1,7,10 The pathogenesis of these syndromes is complex and incompletely understood. The initiating factor in tHUS is endothelial damage by Shiga toxins while it seems to be platelet aggregation in TTP.1,7,10 In recent years, complement hyperactivation has been proposed as an important common pathogenetic factor, due to its prothrombotic and proinflammatory effects (platelets are activated by complement); the glomerular endothelium, which is targeted in aHUS, is highly susceptible to complement. In tHUS, P-selectin upregulation in endothelial cells by Shiga toxins results in complement deposition on these. Hyperactivation of the alternative complement pathway is central to aHUS pathogenesis. In TTP, the large platelet thrombi may contribute to complement activation. This hypothesis is supported by the clinical efficiency of an anti-C5 antibody in aHUS and some cases of tHUS and TTP.10
The pathology of tHUS, aHUS and TTP is essentially similar. Microscopic hallmarks of the acute phase of these conditions in the kidney are: 1) occlusion of glomerular capillaries by platelet-rich fibrin thrombi, 2) capillary wall thickening due to endothelial cell swelling and subendothelial cell debris and fibrin deposits, 3) mesangial damage (cells and matrix), and 4) often fibrinoid necrosis of interlobular and afferent glomerular arterioles. These vascular lesions may lead to variably extensive cortical necrosis. Chronic lesions are only seen in aHUS and TTP.1
Hemolytic-uremic syndrome is better known in human medicine, but natural cases have been reported in horses and dogs;2,3,4,6,8,9 the cause was not determined except in one equine case, involving a mare and her foal, in which a 0138:H2 STEC was isolated from the mare’s uterus and both animal’s gastrointestinal tract.4
JPC Diagnosis: Kidney, cortex: Necrosis and hemorrhage, diffuse, severe with fibrinoid vascular necrosis and numerous fibrin thrombi.
Conference Comment: The contributor has provided an excellent review for hemolytic-uremic syndrome in humans. As previously discussed, “typical” hemolytic-uremic syndrome may result from endothelial damage by Shiga toxins produced by several strains of enteropathogenic E. coli (EPEC) This toxin, encoded in the genome of bacteriophages in these strains, binds to the glycolipid receptor globotriaosylcermaide (Gb3) on the surface of endothelial cells.13 A number of mechanisms result in necrosis of infected cells, including the inhibition of protein synthesis, leading to vascular permeability and cell lysis. The concomitant exposure of endothelial cells to lipopolysaccharides or various inflammatory cytokines (TN F–alpha, IL-1) increases the sensitivity of endothelial cells to the cytotoxic effects of Shiga toxin.
The susceptibility of a particular cell or tissue to the effects of Shiga toxin is directly proportional to the concentration of the Gb3 receptor in a particular tissue. The classic lesions of edema disease in in swine, a disease associated with enterohemorrhagic O serotypes of E., coli (O138, O139, O140)– edema of the eyelids, gastric wall, mesocolon, larynx, and cerebrum – are directly related to the high concentrations of Gb3 receptors in these tissues.
The most widely recognized Shiga-toxin producing E. coli (STEC) is 0157: H7, a major human pathogen, although over 200 other STEC serotypes have been identified.13 In calves less than four weeks of age, STEC have been associated with ulcerative fibrinohemorrhagic enterocolitis and dysentery. Lesions are most commonly seen in the spiral colon and rectum, although occasionally the ileum and cecum are involved. In a review of outbreaks of human enterohemorrhagic E. coli (EHEC), cattle were identified as a natural reservoir of STEC and approximately 75% of outbreaks in humans are limited to the consumption of contaminated beef.11
In dogs, STEC have been associated with dysentery and in greyhounds, the syndrome of cutaneous and renal glomerular vasculopathy has been attributed to the consumption of STEC-contaminated beef.13 A similar condition of cutaneous and renal glomerular vasculopathy has been recently identified in the UK in dogs, although STEC were not identified in feces from affected dogs.5
Another very interesting lesion resembling those caused by STEC has been identified in the mesentery of horses with naturally and experimental endotoxemia. The lesion consists of marked necrosis and loss of medial smooth muscle cells in the mesenteric arterioles with fibrinoid degeneration and intramural hemorrhage.12
The moderator discussed thrombotic microangiopathy, a term which covers a wide range of conditions (infectious disease, drug reactions, and hypertension among others) in which there is direct damage to endothelial cells. The morphology of renal damage among the disparate forms of TMA does not always target the same vessels in the kidney, as “not all endothelial cells are alike.” While the large areas of infarction are the overwhelming feature of this particular slide, careful inspection of will reveal inflammatory cells and cellular debris within the walls of radial arteries ( a change referred to in some quarters as “necrotizing arteritis of Weisbrode”) as well as the present of occlusive thrombi; thrombi are also present in glomerular tuft capillaries as well. The possibility of purpura hemorrhagica as an etiology (which would cause a lesion more similar to TTP than HUS) was also discussed in this case.
While the clinical picture and lesion morphology is consistent with documented cases of hemolytic-uremic syndrome in the horse, the unavailability of hematologic data, coupled with the inability to identify Shiga toxins (and only a section of kidney to review) may call for a less specific diagnosis than hemolytic-uremia syndrome in this particular case.
Contributing Institution:
Université de Montréal, Faculty of veterinary medicine, St-Hyacinthe, Quebec, Canada. http://www.medvet.umontreal.ca
References:
- Alpers CE. The kidney. In: Robbins and Cotran Pathologic Basis of Disease. 8th ed. Philadelphia: Saunders Elsevier; 2010:905-969.
- Chantrey J, Chapman PS, Patterson-Kane. Haemolytic-uraemic syndrome in a dog. J Vet Med A. 2002;49:470-472.
- Dell’Orco M, Bertazzolo W, Pagliaro L et al. Hemolytic-uremic syndrome in a dog. Vet Clin Pathol. 2005;34(3): 264-269.
- Dickinson CE, Gould DH, Davidson AH et al. Hemolytic-uremic syndrome in a postpartum mare concurrent with encephalopathy in the neonatal foal. J Vet Diagn Invest. 2008;20:239-242.
- Holm, LP, Hawkins I, Robin C, Newton RJ, Stanzani G, McMahon LA, Pesavento P, Carr T, Cogen T, Gouto CG, Cianciolo R, Walker $. Cutaneous and renal glomerular vasculopathy as a cause of acute kidney injury in dogs in the UK. Vet Rec 2015; 176(15):384
- Holloway S, Senior D, Roth L et al. Hemolytic uremic syndrome in dogs. J Vet Intern Med. 1993; 7(4):220-227.
- Kumar V, Abbas AK, Fausto N et al. Diseases of white blood cells, lymph nodes, spleen, and thymus. In: Robbins and Cotran Pathologic Basis of Disease. 8th ed. Philadelphia: Saunders Elsevier; 2010:589-638.
- MacLachlan NJ, Divers TJ. Hemolytic anemia and fibrinoid change of renal vessels in a horse. J Am Vet Med Assoc. 1982 Oct 1;181(7):716-717.
- Morris CF, Robertson JL, Mann PC et al. Hemolytic uremic-like syndrome in two horses. J Am Vet Med Assoc. 1987 Dec 1;191(11):1453-1454.
10 Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 2012 Nov;8(11):622-33.
- Nguyen Y, Sperandio V. Enterohemorrhagic E. coli pathogenesis. Front Cell Infect Micro 2012; 2:1-7.
- Oikawa M, Ueno T, Yoshikawa H. Arterionecrosis of the equine mesentery in naturally occurring endotoxaemia. J Comp Path 2004; 150-75-79.
- Uzal FA, Plattner BL, Hosstetter JM. Alimentary System. In: Maxie MG, ed. Jubb Kennedy and Palmer’s Pathology of Domestic Animals, 6th ed. 2016; St. Louis MO, Elsevier Press, vol 2, p. 162-163.