Signalment:
Gross Description:
Histopathologic Description:
Other lesions included erythroid extramedullary hematopoiesis of the spleen, follicular hyperplasia of the splenic white pulp, multifocal nonsuppurative myocar-ditis, multifocal centrilobular hepatic lipidosis, and hemosiderophages in the liver, spleen, blood vessels, alveolar walls, and nasolacrimal ducts.
The granular material does not polarize and does not lose color when bleached.
Special stains: Puchtler-Sweat and Perls [Iron stains]: Blue granules were seen within tubular epithelium but not associated with the basement membrane. No staining was present in the glomeruli.
Massons trichrome (-); PAS (-); and von Kossa and Alizarin Red (-).
Transmission Electron Microscopy:
Kidney, Glomerulus: Within the podocytes, there are 150-750 nm granular deposits adjacent to the basement membrane (white arrow). Similar but smaller deposits are seen within the basement membrane (black arrow). The foot processes are effaced with infrequent filter slits. The mesangium is moderately expanded without an increase in cellularity.
br> Kidney, Tubule: Small granules (<150 nm) are within the basement membrane (black arrow) and adjacent to the basement membrane (white arrow). There is a slight increase in lipid.
Elemental analysis of the granules was attempted but was inconclusive.
Morphologic Diagnosis:
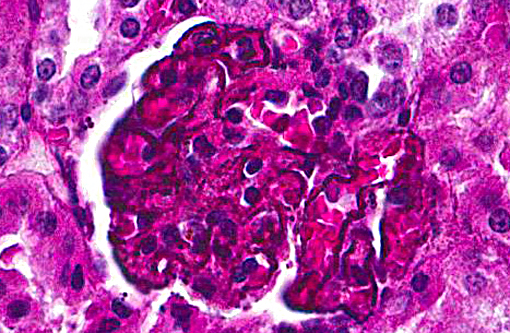
Kidney:glomerulonephropathy characterized by deposition of granular material along [capillary loops] glomerular basement membranes, diffuse, marked.
Kidney, tubules, basement membrane: granular pigmented material deposition.
Kidney: tubular proteinosis, multifocal, mild.
Kidney: chronic interstitial nephritis, multifocal, mild.
Lab Results:
Condition:
Contributor Comment:
If there is a full recovery, there is no progress to chronic kidney disease.(3) Microscopic changes are variable, but tubular degeneration and necrosis predominate. Parasitized erythrocytes are sequestered in glomerular and interstitial vessels, and hemosiderin in tubular epithelium, hemoglobin casts, mild glomerular changes, and interstitial nephritis may also be present. Immune-mediated glomerulonephritis is not associated with acute kidney injury.(3,7) Renal changes may be due to obstruction and sequestration of infected erythrocytes, glomerular and tubular inflammatory changes, fluid loss, and altered renal circulation.(3)
There have been reports of a quartan malarial nephrotic syndrome due to immune complex disease in children in Nigeria that was thought to be related to Plasmodium malariae infection.(6) Subsequent papers have suggested that renal disease in children in Nigeria, as well as Uganda and Ivory Coast, was associated with, but not caused, by Plasmodium malariae infection.(3,4)
Thicket rats found in sub-Saharan Africa are natural hosts for chronic Plasmodium chabaudi, P. berghei, P. vinckei, and P. yoeliiinfection and model chronic Plasmodium falciparum disease in man. Anopheles mosquitos serve as vectors for the both human and rodent plasmodial infection. Although no one species mimics Plasmodium falciparum infection completely, there are enough similarities, especially with Plasmodium chabaudi, to make them a valuable tool for comparative study. In both Plasmodium chabaudi and Plasmodium falciparum, symptoms develop as parasitemia increases and then resolve when parasitemia decreases. Infected rodents develop anemia secondary to dyserythropoiesis, suppression of hematopoiesis, and thrombocytopenia as seen in man. Red blood cells sequester in the placenta, which serves as a model of malaria-related fetal and neonatal abnormalities. In several ways, Plasmodium chabaudi is not representative of infection by Plasmodium falciparum. Unlike Plasmodium falciparum in man, the organ of sequestration is the liver, and not the brain. Infected animals develop hypothermia instead of fever. Although there is splenomegaly, it is not correlated with level of parasitemia or sequestration.(10)
In a recent report, mice infected with Plasmodia berghei ANKA developed renal failure accompanied by increased inflammatory cytokines, expression of adhesion molecules, and products of oxidation. Resulting changes in the vascular endothelium led to interstitial edema and, over time, mice developed acute tubulointerstitial nephritis. Although there was no deposition of brown granular material apparent by examination of H&E slides, there was deposition of polarizable crystals identified as hemozoin in glomeruli and endothelium.(5)
The degree of pigment deposition in the glomeruli was unusual. Analysis by special stains, electron microscopy, and elemental analysis did not reveal the composition of the granular material. Given the high level of parasitemia, we felt the material was directly related to infection with malaria.
JPC Diagnosis:
1. Kidney, cortex, glomerular and tubular basement membranes: Immune complex deposition, diffuse.
2. Kidney, cortex: Tubular ectasia and proteinosis, multifocal, mild.
3. Kidney: Cortical perivascular lymphocytic infiltrates, multifocal, mild.
Conference Comment:
A definitive diagnosis of immune-mediated glomerulonephropathy requires immunofluorescent or immunohistochemical confirmation of immunoglobulin and complement components in glomerular tufts. Ultrastructurally, electron-dense deposits may be seen in mesangial, subepithelial or subendothelial locations. Glomerulonephritis results following the deposition of immune complexes within the glomeruli or following the formation of antibodies to antigens present in the basement membrane (anti-GBM disease). Immune complex glomerulonephritis is a sequela to persistent infections of various etiologies including viral, bacterial and parasitic infections, as well as autoimmune disease. Antigen-antibody complexes form in the presence of antigen excess or when antigen-antibody quantities are equal. The complexes deposit in glomerular capillaries, fix complement, and cause basement membrane damage, which is enhanced by the ensuing inflammatory response. Factors that can determine the extent of deposition include the size and charge of complexes, glomerular permeability and the antigen-antibody avidity. Small and intermediate complexes are the most damaging, as larger complexes are removed from circulation by the mononuclear phagocytic system.(9)
Glomerular lesions described in human malaria patients do include prominent mesangial proliferation with occasional basement membrane thickening and deposition of an eosinophilic granular material along capillary walls, within the mesangium and in Bowmans capsule.(2,3) The origin/nature of this material is not well described. Other findings include immunofluorescence demonstration of IgM and C3 in mesangial capillary walls, although other studies have found immune complex deposition was not seen in basement membranes or the mesangium of glomeruli and clinical / clinical pathology findings apparently dont necessarily support the presence of immune complex deposits in all cases of acute renal failure related to malaria infection.(3) However, in other reports in the literature glomerulonephritis is described in cases of acute renal failure in malaria infection and a distinction is made between chronic progressive glomerulopathy which occurs in quartan malaria in Africa, versus acute renal failure associated with falciparum malaria in Southeast Asia, India and sub-Saharan Africa.(2) Ultrastructural findings described in human cases include subendothelial and mesangial electron dense deposits with the presence of granular, fibrillary and amorphous material.(2,3) The absence of a prominent glomerular inflammatory response in this case leads to speculation regarding the role of the glomerular deposits in renal function.
In this case, diffusely and globally within glomeruli there are 1-2um, prominent, eosinophilic globules lining capillaries and slight increase in cellularity within the mesangium. There are multifocal glomerular synechia, and the parietal epithelium is mildly hypertrophied. The eosinophilic material is also seen within the basement membrane of adjacent tubules, and there is tubular ectasis, proteinosis and attenuation of tubular epithelium with few areas of tubular epithelial degeneration and necrosis. During the conference, there was discussion regarding the origin of the eosinophilic globules, including calcium, hemosiderin, lipofuscin, and melanin. A battery of histochemical stains performed by the contributor did not shed further light as to the nature the basement membrane deposits.
Nephrotic syndrome is well-documented in human malarial infection, and various histologic patterns of glomerular lesions are reported in the literature, including focal segmental glomerulosclerosis (FSGS), membranoproliferative glomerulonephritis, minimal change disease, and membranous glomerulonephritis, among others [Asinobi AO 2015]. In a 1967 study of the histologic features of nephrotic syndrome in 77 people with quartan malaria (infection with Plasmodium malaria), approximately half had detectable malarial organisms, and all but one had glomerulonephritis, including nine cases histological classified as membranous glomerulonephritis [Kibu-kamusoke JW-1967], which corresponds with the preferred diagnosis offered by Dr. Rachel Cianciolo in this case. In the same study, 63% of the patients were children, thus presumably non-immune, and had a higher incidence of parasitemia as compared to adults. A more recent review article on the topic provided by the contributor summarizes the clinicopathologic findings of malaria-induced renal damage in people, including the various forms of malarial nephropathy and their association with a specific malarial etiology and the patient anamnesis (4). General observations in the review included the finding that non-immune (na+â-»ve) patients have a higher risk of developing acute renal failure than semi-immune people (those living in endemic areas); in quartan malaria (Plasmodium malaria) cases originating from Nigeria and the Ivory Coast, the primary glomerular lesion with light microscopic examination was characterized as membranous nephronpathy; and fatal acute renal failure occurs in cases of falciparum malaria in up to 40% of non-immune patients with high parasitemia above 5% of parasitized erythrocytes.
During the conference, the moderator disclosed that this thicket rat was one of several in an experimental group that either died or was euthanized with similar clinical and histopathologic findings as the one presented in this case. The glomerular lesions, gross findings indicative of nephrotic syndrome (anasarca and pleural effusion), and the clinical history of an immunologically na+â-»ve animal with a high parasitemia lends to the possibility of an emerging animal model for malarial acute nephrotic syndrome/renal failure. Interestingly, glomerulonephritis is described in both rat and mouse models infected with variants of Plasmodium berghei, with primarily mesangial deposition of immuneglobulins (e.g. IgM, IgG, C3, among others) and malarial antigen. [George CR 1976; Ehrich JH 1981). In the rat, the renal lesions were transitory and appeared to resolve within one to three months, unlike the lethal course of disease that occurred in this thicket rat.
References:
1. Autino B, Corbett Y, Castelli F, et al. Pathogenesis of malaria in tissues and blood. Mediterr J Hematol Infect Dis. 2012; 4(1):e2012061.
2. Barsoum RS. Malarial acute renal failure. J Am Soc Nephrol. 2000; 11:2147-2154.
3. Das BS. Renal failure in malaria. J Vector Borne Dis. 2008; 45:83-97.
4. Ehrich JHH, Eke FU. Malaria-induced renal damage: facts and myths. Pediatr Nephrol. 2007; 22(5):626-37.
5. Elias RM, Correa-Costa M, Claudiene RB, et al. Stress and Modification of Renal Vascular Permeability Are Associated with Acute Kidney Injury During P. berghei ANKA Infection. PLOS One. 2012; 7(8):e44004.
6. Hendrickse RG, Adeniyi A, Eddington GM, et al. Quartum Malarial Nephrotic Syndrome. Lancet 1. 1972; 1(7761):11431149.
7. McAdam AJ, Sharpe AH. Infectious diseases. In Kumar V, Abbas A K, Fausto N eds. Robins and Cotran Pathological Basis of Disease. 8th ed. Philadelphia, PA: Elsevier Saunders: 2010:386-388.
8. Mishra SK, Das BS. Malaria and acute kidney injury. Semin Nephrol. 2008; 28(4):395-408.
9. Newman SJ. The Urinary System. In: McGavin MD, Zachary JF, eds. Pathologic Basis of Veterinary Disease. 5th ed. St. Louis, MO: Mosby Elsevier; 2012:620-622.
10. Stephens R, Culleton RL, Lamb TJ. The contribution of Plasmodium chabaudi to our understanding of malaria. Trends Parasitol. 2012; 28(2):73-82.
11. Schwartz DA, Genta RM, Bennett, DP, Pomerantz RJ. Infectious and parasitic diseases. In Rubin R, Strayer DS eds. Rubins Pathology. 5th ed. Baltimore, MD: Lippincott Williams & Wilkins: 8:359-362.
12. World Health Organization. World Malaria Report. 2014:2-3.