Signalment:
Gross Description:
Histopathologic Description:
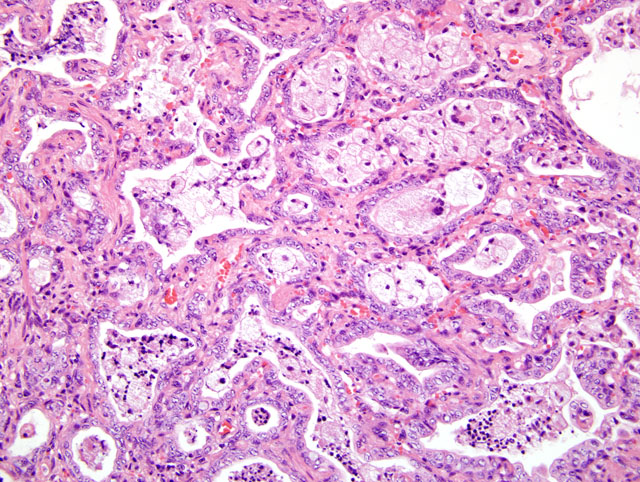
Approximately 70-80% of the lung parenchyma is remodeled by multifocal to coalescing areas of increased cellularity, predominantly located subpleurally, but also extending into the central parenchyma. In these areas, the pulmonary interstitium is markedly expanded by fusiform and plump mesenchymal cells (fibrocytes and fibroblasts), and thick bundles of plump spindloid cells with moderate amounts of eosinophilic cytoplasm, and a medium sized oval to elongated nucleus with a reticular chromatin pattern (myofibroblasts) (Fig. 4-1). Where present, alveolar spaces are lined by a single to occasionally multiple layers of low columnar to cuboidal epithelial cells with indistinct cell borders, a moderate amount of pale eosinophilic, finely granular cytoplasm and a basally located, round to oval nucleus with a finely stippled chromatin pattern, and one to two medium sized nucleoli consistent with type II pneumocytes (honeycomb lung).Â
Within the alveolar lumen are numerous, plump, polyhedral to round cells with abundant, highly vacuolated, pale eosinophilic cytoplasm and a medium sized oval nucleus with a finely stippled chromatin pattern and a single, medium sized nucleolus (alveolar macrophages). These cells are occasionally binucleated and multinucleated. Small aggregates of lymphocytes and plasma cells are scattered throughout the remaining pulmonary parenchyma.Â
Small to medium sized bronchi contain numerous alveolar macrophages interspersed with abundant pale amphophilic to basophilic, lightly fibrillar, acellular material (mucus), small areas of deeply basophilic, cracked acellular material (mineral) and acicular (cholesterol) clefts. At the periphery of the large bronchi are numerous, small peri-bronchiolar glands. In less severely affected areas, the alveolar lumens are multifocally enlarged by non-staining spaces (emphysema).Â
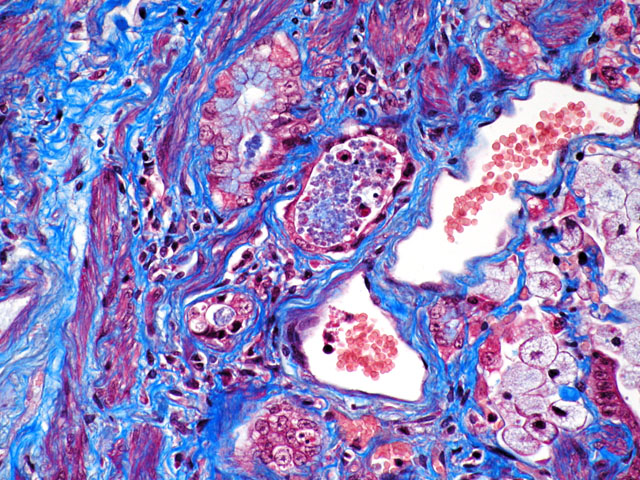
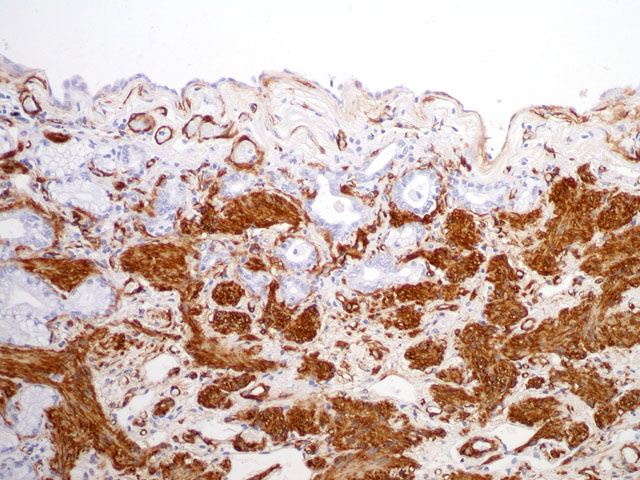
Special stains/Immunohistochemistry: The cuboidal cells lining the alveolae are strongly positive for cytokeratin, confirming their epithelial nature. The interstitial mesenchymal proliferation stained variably blue and pink (collagen and smooth muscle, respectively) (Fig. 4-2) and large areas stained strongly with smooth muscle actin confirming the presence of smooth muscle (Fig. 4-3).
Morphologic Diagnosis:
Condition:
Contributor Comment:
FIPF is an uncommon condition, first described in 2000 by Rhind and Gunn-Moore(2). The average age of onset is approximately 8.7 years and the average time between onset clinical signs to death is 5.5 months.(1) This case has many features characteristic of this condition: sub-plural and caudo-dorsal distribution, persistent progressive fibrosis with temporal heterogeneity of lesions, alveolar type II pneumocyte hyperplasia and small foci of alveolar epithelial squamous metaplasia in some sections. This condition has been associated with the development of bronchoalveolar carcinoma, which was not a feature of this case. The aetiology is not currently known. Toxic, hypersensitivity, inflammatory and genetic origins have been investigated.Â
Parallels have been drawn between FIPF and Usual Interstitial Pneumonia (UIP) in man.(3) Salient features of UIP in man include interstitial fibrosis with fibroblast/ myofibroblast proliferation, temporal heterogeneity of lung remodeling, subpleural orientation, honeycomb change, interstitial smooth muscle proliferation and scant inflammation.
A type II pneumocyte defect has been described in a familial form of human interstitial pulmonary fibrosis (IPF), with abnormal cytoplasmic lamellar body-like inclusions seen on electron microscopy.(3) Similar ultrastructural changes were observed in type II pneumocytes of affected cats by Williams et al. The authors suggested that FIPF may be due to a defect in the type II pneumocyte and that cats potentially represent a spontaneous model of IPF in humans.Â
JPC Diagnosis:
Conference Comment:
References:
2. Rhind S and Gunn-Moore D: Desquamative form of Cryptogenic Fibrosing Alveolitis in a cat. J Comp Path 123:226-229, 2000
3. Williams K, Malarkey D, Cohn L, Patrick D, Dye J and Toews G: Identification of Spontaneous Feline Idiopathic Pulmonary Fibrosis. Chest 125:2278-2288, 2004