Results
AFIP Wednesday Slide Conference - No. 19
- February 3, 1999
-
- Conference Moderator:
Dr. Michael A. Eckhaus, Diplomate, ACVP
NCRR LSS SSB, Bldg. 28A, Room 117
28 Library Drive, MSC 5210
- Bethesda, MD 20892-5210
-
- NOTE: Click on images for larger views. Use
browser's "Back" button to return to this page.
Return to WSC Case Menu
-
Case I - OL8115 (AFIP 2648175)
- Signalment: Ten-week-old, Brown Norway rats.
-
- History: The rats were quality-control animals. They
showed no clinical signs.
- Gross Pathology: There were multiple, small, brown to red
foci over the pleural surface of all lung lobes
Laboratory Results:
- 1. Serology: Negative.
2. Parasitology: Negative.
3. Microbiology: Staphylococcus aureus, Staphylococcus xylosus,
Streptococcus sanguis, and Streptococcus bovis were recovered
from the nasopharynx.
-
- Contributor's Diagnosis and Comments: Lung: Peribronchiolar
and interstitial pneumonia, granulomatous and eosinophilic, multifocal
(varied severity among sections).
Etiology: Unknown.
Multifocally throughout the lung there are variably-sized, discrete,
noncaseating granulomas, mostly arranged around bronchioles,
but also present around or adjacent to vessels in the interstitium.
Granulomas are usually composed of tightly packed epithelioid
macrophages admixed with frequent multinucleated, Langhans-type
and foreign body-type giant cells and mild to moderate numbers
of eosinophils and neutrophils. Within granulomas, there are
occasional small central areas of necrosis characterized by accumulation
of cellular and nuclear debris, degenerate polymorphonuclear
leukocytes, and rarely, deposition of small amounts of brightly
eosinophilic, club-shaped, amorphous material (Splendore-Hoeppli
material; not present in all sections). In addition, throughout
the interstitium there are small to conspicuous perivascular
cuffs composed mostly of eosinophils and neutrophils, admixed
with occasional lymphocytes. In association with inflammatory
infiltrates, there is also mild to moderate alveolar histiocytosis,
mild to moderate interstitial edema, mild thickening of alveolar
walls with type II pneumocyte hyperplasia, and mild to moderate
hyperplasia of bronchiolar epithelia with mucous metaplasia.
-
- Multifocal granulomatous pneumonia is a condition affecting
Brown Norway rats. This condition has been observed worldwide,
but its exact incidence is not known. The only published information
reported this condition in 11 of 12, 10-week-old, Brown Norway
rats, and in 7 of 10 retired breeders (1). Based on macroscopic
examination, the condition can be diagnosed in approximately
25% of 8 to 10-week-old Brown Norway rats. Females appear more
frequently affected than males, and the incidence in retired
breeders is lower, suggesting that this condition may regress
(Charles Clifford, Charles River Laboratories, personal communication).
The pulmonary lesions are not associated with clinical signs
and do not affect the lifespan of the rats.
-
- Grossly, multiple gray to gray-brown foci can be observed
over the pleural surface of all pulmonary lobes. Histologically,
the condition is characterized by multifocal dense aggregates
of mostly macrophages admixed with frequent multinucleated giant
cells, eosinophils, and neutrophils, with fewer lymphoid cells.
The inflammatory infiltrates are peribronchiolar and interstitial,
but usually not in airway lumens.
-
- The cause of this condition is unknown. Serology, microbiologic
tests and special stains (Warthin-Starry, acid-fast, Gram) do
not demonstrate an infectious agent. In addition, the condition
does not appear to be contagious, as rats of other strains housed
in the same rooms do not develop the pulmonary lesions. Recently,
pulmonary inflammatory lesions of unknown etiology have been
reported in Fischer 344 rats used in chronic toxicity studies
(2). These lesions were similar to those of Brown Norway rats
in that they were more commonly observed in younger animals.
However, the inflammatory infiltrates in Fischer 344 animals
were mostly lymphocytic, in contrast to the histiocytic infiltrates
of Brown Norway rats suggesting that these two conditions are
different. Alternatively, the different nature of the inflammatory
infiltrates may simply reflect strain differences in inflammatory
response (3,6).
-
- In humans, multiple types of noninfectious granulomatous
pulmonary conditions of unknown etiology are recognized, including
sarcoidosis, Wegener's granulomatosis, Histiocytosis X (pulmonary
Langerhans cell granulomatosis), and hypersensitivity pneumonitis
(4,5,7). There is some overlap in the type, distribution, and
histologic appearance of pulmonary lesions among these disorders.
The exact cause of these conditions is unknown, but is most likely
related to an uncontrolled response of the immune system (4,5,7).
-
- Brown Norway rats have been used as models of allergic respiratory
disease such as asthma, because of their high capacity for IgE
production and their airway hyperresponsiveness following exposure
to allergens (such as ovalbumin) or some chemicals (1). In addition,
this strain is known for the development of autoimmune syndromes
following administration of mercuric chloride, gold, and penicillamine
(6). Compared to other strains of rats, Brown Norway rats have
low numbers of CD8+ T cells, CD8+ CD45RChigh, and CD4+ CD45RChigh
(6). Because of their unique immune system and the histologic
nature of the lesions, it is tempting to speculate that this
granulomatous pneumonia may result from a disordered immune response
to an unknown antigen. Recently, small, noncaseating granulomas
containing giant cells have been observed in the lungs of Brown
Norway rats following mercuric chloride administration (6). The
lesions described were very similar to this idiopathic granulomatous
pneumonia of Brown Norway rats. This finding suggests that this
common background lesion may represent a confounding factor for
the interpretation of studies involving Brown Norway rats.
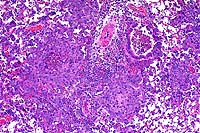 10x
obj
10x
obj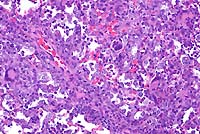 20x
obj
20x
obj
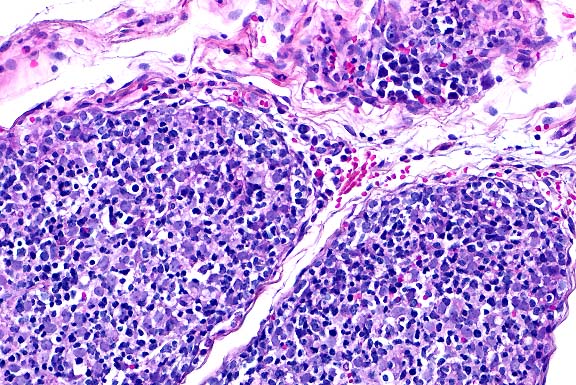
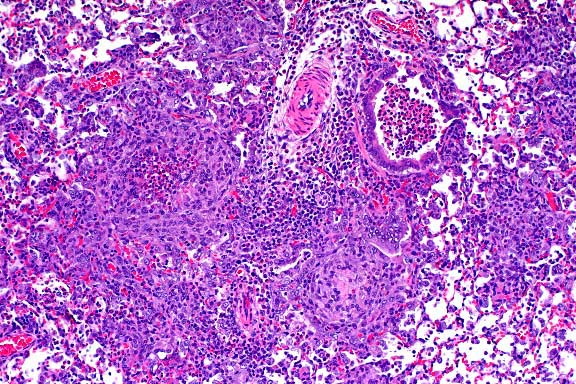
- Case 19-1. Lung. Multifocally throughout the lung,
abundant macrophages, eosinophils, neutrophils and scattered
foreign body or Langhans giant cells expand or replace alveoli
and bronchioles.
-
- AFIP Diagnosis: Lung: Pneumonia, granulomatous and
eosinophilic, peribronchiolar and perivascular, multifocal, moderate,
with perivascular edema, Brown Norway rat, rodent.
-
- Conference Note: Like the contributor, participants
identified perivascular and peribronchiolar inflammatory infiltrates
composed of macrophages with multinucleate giant cells and eosinophils,
and fewer lymphocytes and plasma cells. Inflammatory cells expand
adjacent alveolar septa, and sometimes obscure or fill alveoli,
but are rarely found within bronchiolar airways. Various histochemical
stains performed at the AFIP (tissue Gram stains, acid-fast stains,
periodic-acid Schiff reaction, and Grocott's methenamine silver
method) did not demonstrate the presence of an infectious agent.
-
- The contributor noted the recent report of perivascular inflammatory
lesions in the lungs of Fischer 344 rats composed predominately
of lymphocytes with an associated alveolar exudate of macrophages,
neutrophils, and lymphocytes. An increase in peribronchiolar
lymphoid tissue was also observed. More recently, pulmonary inflammatory
lesions with histomorphologic features similar to those in F344
rats were described in Wistar rats.
-
- While still of unknown etiology, several common features
suggest that these strain-specific lesions may have a similar
pathogenesis: lack of clinical signs in affected animals, distribution
of inflammatory infiltrates, presence of more extensive inflammatory
lesions in young animals, and age-related spontaneous regression
of lesions. While the character of the inflammation in the Brown
Norway rat is different from the other two strains, i.e. granulomatous
and eosinophilic versus lymphocytic, this may be attributed to
variation in immune response as suggested by the contributor.
- The etiology of the inflammatory lesions was not determined
in the Brown Norway or Wistar rats. In the report of F344 rats,
variable (often small) numbers of rod-shaped bacteria were observed
ultrastructurally within macrophages or degenerate cells in alveoli
of most animals with lung lesions.
- Contributor: Searle, 4901 Searle Parkway, Skokie,
IL 60077.
-
- References:
- 1. Ohtsuka R, Doi K, Itagaki S: Histological characteristics
of respiratory system in Brown Norway rat. Exp Anim 46:127-133,
1997.
- 2. Elwell MR, Mahler JF, Rao GN: Inflammatory lesions in
the lungs of rats. Toxicol Pathol 25:529-531, 1997.
- 3. Sorden SD and Castleman WL: Brown Norway rats are high
responders to bronchiolitis, pneumonia, and bronchiolar mastocytosis
induced by parainfluenza virus. Exp Lung Res 17:1025-1045, 1991.
- 4. Jones WW, Geraint JD: Pulmonary Langerhans' cell granulomatosis
(LCG). Sarcoidosis 10:104-107, 1993.
- 5. Soler P, Tazi A, Hance AJ: Pulmonary Langerhans cell granulomatosis.
Cur Opi Pulmon Med 1:406-416, 1995.
- 6. Qasim FJ, Thiru S, Mathieson PW, Oliveira DB: The time
course and characterization of mercuric chloride-induced immunopathology
in the Brown Norway rat. J Autoimmun 8:193-208, 1995.
- 7. Fleming MV, Travis WD: Interstitial lung disease. Pathol
4:121, 1996.
- 8. Slaoui M, Dreef C, van Esch E: Inflammatory lesions in
the lungs of Wistar rats. Toxicol Pathol 26:712-713, 1998.
-
Case II - 1971508 (AFIP 2642673)
- Signalment: Seven-week-old, Jack Russell terriers,
canine.
-
- History: Seven 7-week-old puppies were examined because
of a history of unexplained mortality in previous litters. Six
of 14 puppies from three litters of the same breeding pair died
unexpectedly at eight weeks of age shortly after vaccination.
Administration of modified live virus vaccines produced hepatic
necrosis with inclusions, characteristic of adenoviral hepatitis.
In this case, four unvaccinated puppies from a litter of seven
were chosen based on the finding of lymphopenia. There were no
significant clinical signs in any of the animals. These pups
were examined at necropsy, along with two non-lymphopenic littermates.
-
- Gross Pathology: In four of the six puppies necropsied,
the thymus was markedly hypoplastic in appearance and weighed
from 0.5 to 2 grams. In contrast, the two non-lymphopenic litter
mates had thymic weights of 8.5 and 10 grams. Hypoplasia of lymph
nodes and hyposplenism was also noted in the four lymphopenic
puppies. Other gross lesions in the lymphopenic pups included
diffuse, mild catarrhal enterocolitis and, in one pup, mild,
acute multifocal necrotizing hepatitis. All six animals were
severely flea infested, but had no internal parasites.
-
- Laboratory Results: The four animals with reduced
thymic weight had leukopenia with severe relative and absolute
lymphopenia (an average of 2% lymphocytes ranging from 0-5% of
the total WBC with an absolute count of 0.1 x 103/ml ranging
from 0-0.2 x 103/ml), a mild relative neutrophilia, and severely
reduced IgM (undetected with all values less than 10 mg/100 ml
in the four lymphopenic animals compared with non-lymphopenic
litter mates having a mean of 173 mg/100 ml ranging from 140-200
mg/100 ml) and an IgG (mean of 115 mg/100 ml ranging from 60-200
mg/100 ml compared with non-lymphopenic litter mates having a
mean of 417 mg/100 ml ranging from 350-500 mg/100 ml). All animals
had a moderate regenerative anemia and mild monocytosis. Platelet
counts were normal. Previous litters had no blood count data,
but a fourteen-week-old, unvaccinated pup had undetectable IgA,
IgG and IgM a few days before death.
-
- Contributor's Diagnoses and Comments:
- 1. Marked thymic cortical hypoplasia.
- 2. Severe combined immunodeficiency (SCID), Jack Russell
terrier.
-
- The thymic tissues presented for examination represent age
and sex-matched controls of affected versus unaffected littermates.
Affected puppies had markedly diminished thymic mass, with primary
depletion of thymic cortical elements. Lymphoid hypoplasia was
also present (tissues not submitted) in the spleen (no periarteriolar
sheaths or follicles), lymph nodes, and GALT/BALT. The immunophenotypic
analysis of lymphocytes by cluster of differentiation (CD) typing
is not yet available. The remarkable paucity of circulating lymphocytes
did not afford the opportunity for in vitro mitogen stimulation.
-
- The marked lymphoid hypoplasia and insignificant amounts
of IgM in seven-week-old animals, coupled with a history of death
at the time of maternal antibody titer loss, strongly suggests
a novel, severe combined immunodeficiency (SCID) of Jack Russell
terriers. Further, the one to one female to male ratio among
10 affected animals of 24 at risk with the same, clinically normal
sire and dam suggests an autosomal recessive trait. Acquired
immunodeficiencies occur in the canine, but are usually characterized
by incomplete lymphopenia, deficiencies of single or multiple
components of the immune system, and often a marked proliferative
response in phagocytic (reticuloendothelial) systems.
- Canine severe combined immunodeficiency has been reported
in the Basset Hound and Cardigan Welsh Corgi breeds to date.
In these breeds, the disease is inherited as an X-linked recessive
trait (XSCID), and thus affects only male puppies. The disease
has been determined to be due to a four-nucleotide deletion defect
in the genetic locus coding for the gamma chain of the interleukin-2
receptor (IL-2R).1 Affected puppies have increased proportions
of immature thymocytes, normal relative numbers of B-lymphocytes,
and normal IgM levels, although T-lymphocytes (particularly the
CD8+ subset), IgG, and IgA percentages tend to be reduced or
are absent.
-
- In the human being, forms of severe combined immunodeficiency
include an adenosine deaminase deficiency, a defect in RAG-1
or RAG-2, and reticular dysgenesis (may be X-linked inheritance
with deficient T-cells and normal or high levels of B-cells,
or autosomal recessive inheritance with T-cells and B-cells both
deficient).3,4
The most common form of human SCID is also X-linked recessive
in inheritance, and is similar to the disease previously reported
in canines in that it involves a defect in the IL-2R gamma chain
locus. The second most prevalent form of human SCID involves
an autosomal recessive inheritance pattern-based defect in production
of the purine nucleoside metabolizing enzyme, adenosine deaminase
(ADA).4 Defects in the production of the purine metabolizing
enzyme purine nucleoside phosphorylase (PNP) and defects in expression
of MHC class II account for some of the remaining heritable human
SCID cases, though these conditions are much more rare than are
human XSCID and ADA deficiencies.4,5
-
- The disease present in this litter of Jack Russell terriers
represents the first description of non-X-linked SCID in a canine
breed. Both male and female puppies in this litter were affected,
as evidenced by absence of circulating lymphocytes and lymphoid
hypoplasia in multiple tissues. Additional studies to determine
whether this litter may have deficiencies in one or more purine
catabolizing enzymes such as ADA, similar to the more common
non-X-linked human SCID variants, are being performed at the
time of this submission. Equine SCID, which is most common in
the Arabian breed, is inherited as an autosomal recessive trait.
The defect in equine SCID is an absence of the p350 component
of DNA-dependent protein kinase.6 Such a defect in these dogs
cannot be excluded at this time.
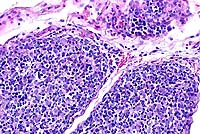 20x
obj
20x
obj
- Case 19-2. Thymus. There is diffuse, marked depletion
of lymphocytes and no distinction between cortical and medullary
zones. Reticular stromal cells remain with few small lymphocytes.
-
- AFIP Diagnoses:
- 1. Thymus and lymph node: Hypoplasia, lymphoid, diffuse,
severe, Jack Russell terrier, canine.
- 2. Thymus and lymph node: Extramedullary hematopoiesis, diffuse,
mild to moderate.
-
- Conference Note: Due to section variation, some slides
may contain only lymph node and no thymus. In the affected thymus,
there is marked decrease in size of thymic lobules, loss of demarcation
between the cortex and medulla, and prominence of medullary epithelial
reticular cells due to marked depletion of lymphocytes. In lymph
node sections, there is marked depletion of lymphocytes, with
virtual absence of paracortical lymphoid cells. Immunohistochemical
staining for CD3, a pantropic T-lymphocyte marker, performed
at the AFIP demonstrated diffuse marked decrease of CD3-positive
cells as compared with the thymus of the age-matched control
littermate, especially within the medulla where mature T-cells
are found. Small numbers of CD3-positive T-cells are present
in the cortex (immature T-lymphocytes).
-
- In horses, SCID is inherited as an autosomal recessive disorder
in Arabian and Appaloosa breeds. Affected foals lack cell-mediated
immunity and are unable to synthesize immunoglobulins. Foals
may remain normal during the first few months of life owing to
colostral transfer of maternally derived IgG, but as passive
immunity wanes, the foals become increasingly susceptible to
infections. The disorder is characterized by lymphopenia, decrease
or absence of immunoglobulins, and death by five months of age.
Death most commonly results from pneumonia caused by adenovirus,
Rhodococcus equi, Pneumocystis carinii, or Cryptosporidium parvum.
The thymus is aplastic or hypoplastic, and the spleen and lymph
nodes lack follicles and contain few lymphocytes and no plasma
cells.
Contributor: Animal Health Diagnostic Laboratory, P.O.
Box 30076, Lansing, MI 48909.
-
- References:
- 1. Pullen RP, Somberg RL, Felsburg PJ, Henthorn PS: X-linked
combined immunodeficiency in a family of Cardigan Welsh Corgis.
J Amer Anim Hosp Assoc 33:494-499, 1997.
- 2. Felsburg PJ, Somberg RL, Perryman LE: Domestic animal
models of severe combined immunodeficiency: Canine X-linked severe
combined immunodeficiency and severe combined immunodeficiency
in horses. Immunodeficiency Reviews 3:277-303, 1992.
- 3. Blaese RM: Genetic immunodeficiency syndromes with defects
in both T and B-lymphocyte function. In: The Metabolic and Molecular
Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle
D, eds., vol. 3, pp. 3895-3909, McGraw-Hill, New York, 1995.
- 4. Hershfield MS, Mitchell BS: Immunodeficiency diseases
caused by adenosine deaminase deficiency and purine nucleoside
phosphorylase deficiency. In: The Metabolic and Molecular Bases
of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle D,
eds., vol. 3, pp. 1725-1768, McGraw-Hill, New York, 1995.
- 5. Rosen FS, Bhan AK: A 54-day-old premature girl with respiratory
distress and persistent pulmonary infiltrates. New Engl J Med
Surg 338:1752-1759, 1998.
- 6. Tizard IR: Primary immune deficiencies. In: Veterinary
Immunology: An Introduction, 5th edition, pp. 445-447, W.B. Saunders,
Philadelphia, PA, 1996.
- 7. Jones TC, Hunt RD, King NW: Immunopathology. In: Veterinary
Pathology, 6th ed., pp. 186-189, Williams and Wilkins, Baltimore,
MD, 1997.
-
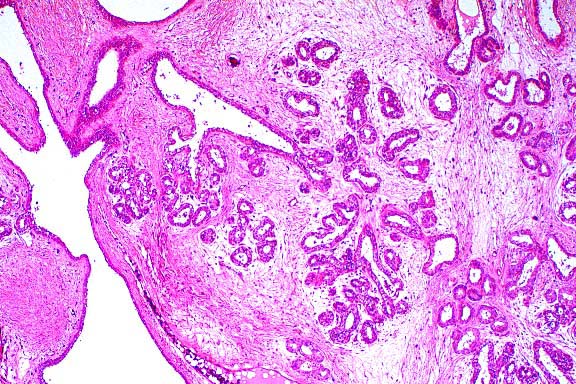
Case III - 98-7050 or 98-7442 (AFIP 2641860)
-
- Signalment:
- 1. 98-7050: 2-month-old, female BALB/cJ-Fechtm1Pas/Fechtm1Pas
mouse.
2. 98-7442: 5-month-old, female BALB/cJ-Fechtm1Pas/Fechtm1Pas
mouse.
Both animals are inbred laboratory mice. The scientific name
is inappropriate, since inbred mice are derived from a variety
of substrains.
-
- History: Both mice were submitted for a limited study.
They were placed under lights in a mouse room to determine if
they routinely devel-oped photosensitization problems. This did
not happen in a conventional mouse room. Ultraviolet lights of
specific wavelengths are required to induce skin lesions. This
was done to address an animal health concern raised by a clinician.
-
- Gross Pathology: Mice submitted were alert and active.
The livers of both mice were firm and dark brown to purple, with
irregular and roughened surfaces. In case 98-7050, the urine
was observed to be orange.
Laboratory Results: None.
- Contributor's Diagnoses and Comments:
- 1. Liver, mild bile duct hyperplasia.
- 2. Liver, moderate biliary fibrosis.
- 3. Liver, mild chronic periportal hepatitis.
4. Liver, porphyrin cholelithiasis.
Etiology: Autosomal recessive mutation, ferrochelatase
deficiency.
-
- Contributor's comments consist of text taken directly
from: Montagutelli X: The ferrochelatase deficiency (Fechm1Pas)
mutation, chromosome 18. In: Handbook of Mouse Mutations with
Skin and Hair Abnormalities, Sundberg JP, ed., pp. 247-251, CRC
Press Inc., Boca Raton, FL, 1994.
- The ferrochelatase deficiency (Fechm1Pas) mutation arose
in 1988 at the Institut Pasteur in Paris in a mutagenesis experiment
with ethylnitrosourea.1 The first features observed were an intense
yellow color of the serum, reduced hematocrit, and grossly evident
jaundice in albino mutant mice. This condition was transmitted
in an autosomal recessive manner. The mutation was introduced,
through several backcrosses, onto the BALB/cByJ inbred background,
where the anomalies appeared to be the most severe.
-
- The influence of genetic background on the development of
jaundice has been observed through the intercrossing of heterozygotes
derived from the cross of homozygous mutants (close to BALB/cByJ)
with (-C57BL/6J X SJL/J)F1 hybrids. This intercross did not yield
affected mice with overt jaundice, even though other biological
parameters, such as elevation of protoporphyrin (see below) were
characteristic for this mutation (Montagutelli, unpublished data).
The ferrochelatase deficiency mutation was mapped to mouse chromosome
18, 40 cM from the centromere (Montagutelli, unpublished).
-
- Gross Lesions: On the BALB/cByJ background, mutant
mice can be recognized as early as two weeks of age by the intense
yellow color of their serum and by gross bilirubinuria. Jaundice
is apparent by the yellow coloration of the unpigmented ears.
Photosensitivity is often observed in homozygotes under standard
husbandry conditions (fluorescent lighting). Inflammatory lesions
appear primarily on the ears, which initially become red and
swollen. In some mice, ear tips undergo necrosis, and multiple
ulcerations develop. Necrotic ear tips undergo autoamputation
which eventually heal, leaving deformed pinnae. Adult homozygotes
have an enlarged abdomen due to marked hepatomegaly and splenomegaly,
which is progressive from the first days of life. On the BALB/cByJ
background, males are usually fertile whereas females breed rarely.
No anomalies have been observed in heterozygotes, other than
a transient and mild jaundice that may be seen at 4 to 5 weeks
of age.
-
- Microscopic Lesions: Very severe liver lesions are
observed in homozygous ferrochelatase deficiency mutant mice.
At 15 days of age there is a 65% increase in the liver:total
body weight ratio. Microscopic examination reveals portal and
periportal fibrosis (Fig. 2) as well as focal accumulation of
dense, dark brown pigment in canaliculi, interlobular biliary
ducts and Kupffer cells (Fig. 3). Erythroid hyperplasia is prominent
in the bone marrow and spleen.
-
- A normocytic anemia develops after one month of age in homozygotes.
At six months of age, hemoglobin concentration, red blood cell
counts, and hematocrits are decreased by 25-30% that of controls
or heterozygotes. Red blood cells are more heterogeneous, as
determined by the increased volume range distribution width and
the heterogeneity of cell resistance to osmotic lysis. Polychromasia,
anisocytosis, target cells, and leptocytes are observed in blood
films.
-
- Immunological and Biochemical Abnormalities: Plasma bilirubin
(mainly conjugated bilirubin) is markedly increased (50 fold)
in homozygotes. Protoporphyrin levels are also considerably elevated
in erythrocytes (25 fold), plasma (20-200 fold), liver (1000
fold) and stool (10 fold). Serum alkaline phosphatase and transaminases
are consistently increased, as a consequence of chronic liver
damage. Enzymatic activity of ferrochelatase in spleen, kidney,
and liver in homozygotes is 3-7% of normal controls and close
to 50% of normal in heterozygotes.
-
- Ferrochelatase is the last enzyme of the heme biosynthesis
pathway that catalyzes the insertion of ferrous iron (Fe2+) into
protoporphyrin.2 The cDNA that encodes for ferrochelatase has
been sequenced.3 In the ferrochelatase deficiency mouse mutation,
a T to A transposition at nucleotide 293 was identified in the
gene coding for the defective enzyme. This transposition led
to a methionine to lysine substitution at position 98 in the
protein M98K.4 In vitro expression of the mutant protein leads
to reduced enzymatic activity, similar to that observed in vivo.
-
- Analogous Human Disease: In humans, erythropoietic protoporphyria
(EPP) is associated with reduced activity of ferrochelatase.2,5
The disease is characterized by cutaneous photosensitivity. A
mild microcytic, hypochromic anemia is observed in a minority
of cases. Fatalities from rapidly progressive liver disease have
been reported in at least 20 patients,2,6 which is an indication
for liver transplantation.7-10 Biochemically, EPP results in
the accumulation of protoporphyrin in erythrocytes, plasma, and
feces. EPP is generally assumed to be an autosomal dominant hereditary
condition,2,11 but it may be inherited, in some cases, in an
autosomal recessive fashion.12-14 Four human mutations have been
described so far.
-
- Analogous Animal Diseases: Ferrochelatase deficiency has
been described in cattle.18 Affected cattle develop cutaneous
lesions after exposure to sunlight but they do not develop anemia
or hepatobiliary disease.19,20 Bovine protoporphyria is transmitted
as an autosomal recessive trait.
- Potential Uses of the Ferrochelatase Deficiency Mutation:
The ferrochelatase deficiency mutation is the first murine genetically
determined model for human EPP. Because of the high incidence
and severity of liver disease in the mouse, which represents
the main complication in the human, this model is likely to become
highly used to investigate the human disease and test new therapies.
Availability of Mice: Mice are currently available as heterozygous
breeding pairs on a limited basis from investigators at the Institut
Pasteur in Paris, France. This mutation has been imported into
the Induced Mutant Resource at The Jackson Laboratory, Bar Harbor,
Maine, USA where it is readily available as a second resource.
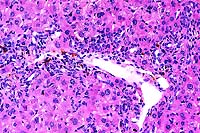 20x
obj
20x
obj
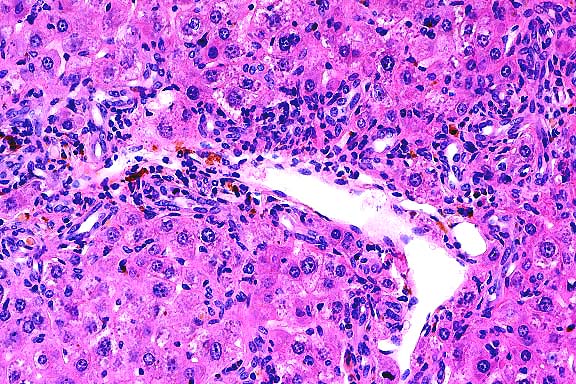
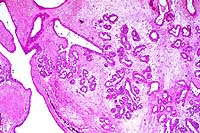
- Case 19-3. Liver. There is diffuse bile duct hyperplasia.
These bile ducts are often surrounded by lymphocytes and plasma
cells. Scattered foci of neutrophils and intrahepatocytic brown
pigment are also present.
-
- AFIP Diagnosis: Liver: Hyperplasia, biliary and oval
cell, portal and periportal, diffuse, moderate, with multifocal
mild lymphocytic and neutrophilic portal and periportal hepatitis,
individual hepatocyte necrosis, and intracellular brown globular
anisotropic pigment, BALB/cJ-Fech mouse, rodent.
-
- Conference Note: Porphyrias are uncommon disorders
caused by disturbances or deficiencies of enzymes involved porphyrin
metabolism. Porphyrins are pigments found in hemoglobin, myoglobin,
and cytochromes. Enzyme deficiencies of heme biosynthesis lead
to excessive accumulation of porphyrins and their precursors.
In erythropoietic porphyrias, enzyme deficiencies may occur during
the formation of protoporphyrin, or, as in this case, at the
last step of heme synthesis. The intermediates that accumulate
during dysfunctional heme synthesis, and the resultant clinical
manifestations, depend upon the step at which the enzymatic defect
occurs.
-
- Congenital erythropoietic porphyrias have been documented
in several domestic animal species, including several breeds
of cattle, swine, and in domestic shorthair and Siamese cats.
The disorder is inherited as an autosomal dominant trait in pigs
and cats, while in cattle it is an autosomal recessive disorder.
Porphyrias resulting from an enzymatic defect of uroporphyrinogen
III cosynthetase, such as in cattle and humans, cause overproduction
of uroporphyrin I, coproporphyrin I, and protoporphyrin III,
which escape the erythrocyte and accumulate in tissues. In cattle,
accumulation of these pigments in bone and dentine leads to pink-red
discoloration, known clinically as osteohemochromatosis and "pink-tooth",
respectively. Anemia occurs due to abnormal hemoglobin production
and decreased erythrocyte life span. Increased renal excretion
of porphyrin imparts an amber-brown discoloration to the urine
(porphyrinuria) which emits a red fluorescence under ultraviolet
light. Type II photodermatitis occurs due to deposition of porphyrins
in the skin.
- Congenital erythropoietic protoporphyria of cattle differs
from bovine congenital porphyria in that affected animals develop
only photodermatitis; discoloration of the teeth and bones, anemia,
and porphyrinuria are not observed. This autosomal recessive
disorder has been described in Limousine cattle, and results
from a deficiency of ferrochelatase leading to accumulation of
protoporphyrin IX in circulation and tissues.
-
- Photodermatitis in animals and humans with erythropoietic
porphyria results from accumulation of photodynamic pigments
in the skin, which absorb ultraviolet and visible light and transform
it into light of longer wavelength (red and infrared). The photoactive
pigments cause production of reactive oxygen metabolites in the
skin, either directly through transfer of energy to oxygen within
the cytosol, or indirectly through activation of xanthine oxidase
by calcium-dependent proteases. Oxygen free radicals then cause
lipid peroxidation of cell membranes, rupture of lysosomes and
mitochondria, complement activation, degranulation of mast cells,
and release of vasoactive factors.
-
- In addition to photodermatitis, variably severe hepatic disease
may occur in human cases of protoporphyria, sometimes manifested
as rapidly progressive liver failure associated with accelerating
photosensitivity and cholestasis. Protoporphyrin IX, an hydrophobic
porphyrin, is cleared from the serum by the liver and secreted
in the bile where it enters the enterohepatic circulation. Cholestasis
results from intracellular and canalicular precipitation of protoporphyrin,
and is associated with inhibition of canalicular sodium/potassium
ATPase. Hepatotoxicity, biliary hyperplasia, and portal fibrosis
result.
-
- Contributor: The Jackson Laboratory, 600 Main Street,
Bar Harbor, ME 04609-1500, and The Institut Pasteur, Unite de
Genetique des Mammiferes, 25 Rue du Docteur Roux, 757244, Paris
Cedex 15, France.
-
- References:
- 1. Montagutelli X: The ferrochelatase deficiency (Fechm1Pas)
mutation, chromosome 18. In: Handbook of Mouse Mutations with
Skin and Hair Abnormalities, Sundberg JP, ed., pp. 247-251, CRC
Press Inc., Boca Raton, FL, 1994.
- 2. Jones TC, Hunt RD, King NW: Mineral deposits and pigments.
In: Veterinary Pathology, 6th ed., pp. 73-75, Williams and Wilkins,
Baltimore, MD, 1997.
- 3. Yager JA, Scott DW: The skin and appendages. In: Pathology
of Domestic Animals, Jubb KVF, Kennedy PC, Palmer N, eds., 4th
ed., vol. 1, pp. 595-596, Academic Press, San Diego, CA, 1993.
- 4. Cotran RS, Kumar V, Collins T: The skin. In: Robbins Pathologic
Basis of Disease, 6th ed., pp. 1205-1206, WB Saunders, Philadelphia,
PA, 1999.
- 5. Cox TM, Graeme JM, Alexander MD, Sarkany RPE: Protoporphyria.
Seminars in Liver Disease 18:85-93, 1998.
-
Case IV - AP#2489 (AFIP 2641824)
- one gross color photo transparency
-
- Signalment: Eleven-month-old, female, Watanabe hyperlipidemic
(WHHL) rabbit.
-
- History: One of four rabbits (three females and one
male) used in a cyclosporine dose response study. The animal
was given cyclosporine (Sandimmune I.V. ä) subcutaneously
once daily at a dosage of 10 mg/kg. Blood cyclo-sporine levels
were monitored once a week, along with blood urea nitrogen and
creatinine levels. Approximately one month after beginning cyclosporine
treatment, all animals began exhibiting signs of decreased food
consumption, mild dehydration, and weight loss. The animal was
euthanized two months following daily cyclosporine treatments.
-
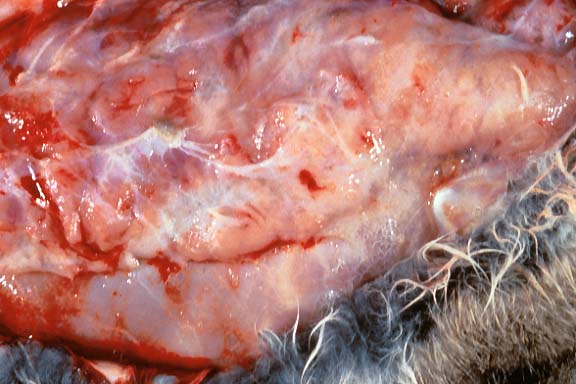
- Gross Pathology: All regions of the mammary gland
were thickened and edematous.
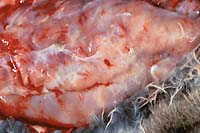
- Case 19-4. Gross Image. This closeup of mammary gland
illustrates the nodularity and high fibrous connective tissue
(white) content of these hyperplastic glands.
-
- Laboratory Results: No abnormalities were detected
in blood urea nitrogen and creatinine levels.
-
- Contributor's Diagnosis and Comments: Hyperplasia,
diffuse, marked, ductal and acinar, mammary gland.
-
- Cyclosporine (cyclosporin A, CsA) is a potent immunosuppres-sive
agent widely used in humans for preventing rejection of organ
transplants and as treatment for autoimmune diseases. Notable
side effects of chronic cyclosporine administration, such as
nephrotoxicity and hepatotoxicity, have been well documented.
Although rare, gynecomastia has been observed in male patients
on cyclosporine therapy and is thought to occur as a result of
an imbalance in the peripheral testosterone-to-estrogen ratio.
This suggests that cyclosporine may cause endocrine dysfunction.
Development of breast fibroadenomas has also been reported in
women treated with cyclosporine.
-
- Little work has been done to elucidate potential side effects
of cyclosporine on endocrine ovarian function. Cyclosporine has
been demonstrated to decrease plasma progesterone levels and
augment the action of follicle stimulating hormone (FSH) in rats
and rabbits. Moreover, studies have shown that the WHHL rabbit
has an abnormal hypothalamic-pituitary-ovarian axis. Normally,
stimulation and growth of the mammary gland is under hormonal
control of both progesterone and estrogen. In the dose response
study from which this case was derived, all females had marked
mammary gland enlargement on post-mortem examination, but similar
lesions were not observed in the male rabbit. Diffuse hyperplasia
of acini and ductules and moderate desmoplasia were observed
in histologic sections. The microscopic lesions strongly suggest
that the inciting cause reflects ovarian hormonal dysfunction
due to long term cyclosporine administration and/or strain characteristics
unique to the WHHL rabbit.
-
- Clinical signs observed during the course of this study (e.g.,
weight loss, decreased appetite) correlate to a toxic syndrome
which has been reported in rabbits treated with cyclosporine.
 4x
obj
4x
obj
- Case 19-4. Mammary gland. Glandular acini are hyperplastic,
multifocally ectatic, and separated by increased amounts of loose
fibrous connective tissue stroma.
-
- AFIP Diagnosis: Mammary gland: Hyperplasia, ductal
and stromal, diffuse, moderate to marked, WHHL rabbit, lagomorph.
-
- Conference Note: In cats, mammary fibroepithelial
hyperplasia, also known as fibroadenoma and fibroadenomatous
hyperplasia, is a condition of young queens, usually less than
two years of age, characterized by benign, nonneoplastic proliferation
of ducts and periductal connective tissue in multiple glands.
The condition also occurs in cats treated with certain medications.
While the condition in cats shares several histologic features
with the mammary gland of this rabbit, the etiology is endogenous
progesterone or exogenous compounds that have progesterone-like
activity; the condition regresses spontaneously following ovariohysterectomy
or discontinuation of medical therapy.
-
- In rabbits, cyclosporine seems to exert a negative effect
on progesterone levels. In an experimental study, the ovaries
of rabbits treated with cyclosporine prior to mating and during
pregnancy contained fewer corpora lutea and had lower serum progesterone
levels than control animals. In rat granulosa cells treated with
cyclosporine in vitro at dosages designed to approximate immunosuppressive
therapy, the drug augmented estrogen production at lower doses
and was inhibitory at higher doses, while progesterone production
was either unaffected or inhibited. These findings suggest that
cyclosporine therapy may directly influence the steroidogenic
function of the ovary, and thus affect the physiological state
of target tissues of the sex steroids, such as the mammary gland;
the exact mechanisms are not completely understood.
-
- The WHHL rabbit has an aberrant hypothalamic-pituitary-ovarian
axis, and it may be difficult to separate the role of this condition
from that of cyclosporine in the pathogenesis of mammary hyperplasia.
This rabbit serves as a model for familial hypercholesterolemia
due to a defect in LDL receptor function which alters cholesterol
availability to cells. Cholesterol is the precursor for steroid
hormone synthesis, and a defective LDL receptor alters steroidogenesis.
The low density lipoprotein particle is especially important
for ovarian steroid synthesis in the corpus luteum, and reduced
availability of cholesterol leads to significant reduction in
plasma progesterone concentrations.
-
- As noted by the contributor, cyclosporine is known for its
potential nephrotoxicity and explains the monitoring of blood
urea nitrogen and creatine levels in these rabbits during the
course of the study. Chronic administration of cyclosporine at
immunosuppressive doses to protect against organ transplant rejection
may cause renal dysfunction due to decreases in glomerular filtration
rates (GFR), decreased renal perfusion, increased secretion of
renin, and activation of the renin-angiotensin system. Decreases
in GFR result from afferent arteriolar degeneration. Lesions
observed in the afferent arterioles consist of endothelial swelling,
medial hyalinosis, and degeneration of the smooth muscle vascular
wall. Very few compounds induce such specific lesions in specialized
blood vessels. Other renal lesions caused by cyclosporine include
vacuolation of the tubular epithelium, cortical interstitial
fibrosis, and perivascular sclerosis of the hilar and interlobular
arteries and arterioles, leading to glomerulosclerosis.
-
- Contributor: Center for Comparative Medicine, Baylor
College of Medicine, One Baylor Plaza, Houston, TX 77030.
-
- References:
- 1. Jacobs U, Klein B, Klehr HU: Cumulative side effects of
cyclosporine and Ca antagonists: Hypergalactinemia, mastadenoma,
and gynecomastia. Transplant Proceedings 26:3122, 1994.
- 2. Rajfer J, Sikka SC, Lemmi C, Koyle MA: Cyclosporine inhibits
testosterone biosynthesis in the rat tes-tis. Endocrinology 121:586-589,
1987.
- 3. Gore-Langton RE: Cyclosporine differentially affects estrogen
and progestin synthesis by rat granulosa cells in vitro. Molecular
and Cellular Endocrinology 57:187-198, 1988.
- 4. Al-Chalabi HA: Effect of cyclosporine A on the morphology
and function of the ovary and fertility in the rabbit. International
Journal of Fertility 29:218-223, 1984.
- 5. Robins ED, Nelson LM, Hoeg JM: Aberrant hypothalamic-pituitary-ovarian
axis in the Watanabe heri-table hyperlipidemic rabbit. Journal
of Lipid Research 35:52-59, 1994.
- 6. Gratwohl A, Riederer I, Graf E, Speck B: Cyclosporine
toxicity in rabbits. Laboratory Animals 20:213-220, 1986.
- 7. Calne RY, et al.: Cyclosporine-A in clinical organ grafting.
Transplantation Proceedings 13:349-358, 1981.
- 8. Robertson JL: Chemically induced glomerular injury: A
review of basic mechanisms and specific xenobiotics. Toxicol
Pathol 26:64-72, 1998.
- 9. Jones TC, Hunt RD, King NW: Genital system. In: Veterinary
Pathology, 6th ed., pp. 1191-1200, Williams and Wilkins, Baltimore,
1997.
-
- Conference Coordinator:
- Ed Stevens, DVM
Captain, United States Army
Registry of Veterinary Pathology*
Department of Veterinary Pathology
Armed Forces Institute of Pathology
(202)782-2615; DSN: 662-2615
Internet: STEVENSE@afip.osd.mil
-
- * The American Veterinary Medical Association and the American
College of Veterinary Pathologists are co-sponsors of the Registry
of Veterinary Pathology. The C.L. Davis Foundation also provides
substantial support for the Registry
- Return to WSC Case Menu
 10x
obj
10x
obj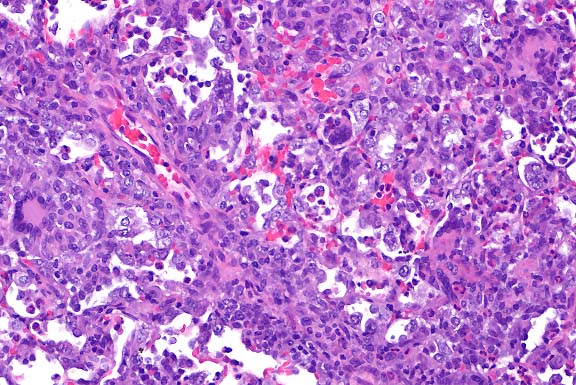 20x
obj
20x
obj